
Discuss evidence referring to the genetic role in congenital heart diseases, whether chromosomic alterations or monogenic diseases.
Data sourceLILACS, PubMed, MEDLINE, SciELO, Google Scholar, and references of the articles found. Review articles, case reports, book chapters, master's theses, and doctoral dissertations were included.
Summary of findingsCongenital heart diseases are among the most common type of birth defects, afflicting up to 1% of the liveborn. Traditionally, the etiology was defined as a multifactorial model, with both genetic and external contribution, and the genetic role was less recognized. Recently, however, as the natural evolution and epidemiology of congenital heart diseases change, the identification of genetic factors has an expanding significance in the clinical and surgical management of syndromic or non‐syndromic heart defects, providing tools for the understanding of heart development.
ConclusionsConcrete knowledge of congenital heart disease etiology and recognition of the genetic alterations may be helpful in the bedside management, defining prognosis and anticipating complications.
Discutir as evidências referentes ao papel genético em cardiopatias congênitas, sejam alterações cromossômicas ou doenças monogênicas.
Fonte de dadosLilacs, PubMed, Medline, SciELO, Google Scholar e referências dos artigos encontrados. Artigos de revisão, relatos de casos, capítulos de livros, dissertações de mestrado e teses de doutorado foram incluídos.
Síntese dos dadosAs cardiopatias congênitas estão entre os tipos mais comuns de defeitos congênitos, afetando até 1% dos nascidos vivos. Tradicionalmente, a etiologia era definida como um modelo multifatorial, com contribuição tanto genética quanto externa, sendo o papel genético menos reconhecido. Recentemente, no entanto, à medida que a evolução natural e a epidemiologia das cardiopatias congênitas mudaram, a identificação de fatores genéticos tem adquirido importância crescente no tratamento clínico e cirúrgico de defeitos cardíacos sindrômicos e não-sindrômicos, fornecendo ferramentas para a compreensão do desenvolvimento do coração.
ConclusõesO conhecimento concreto da etiologia das cardiopatias congênitas e o reconhecimento das alterações genéticas podem ser úteis no tratamento à beira do leito, definindo o prognóstico e antecipando as complicações.
As doenças cardíacas congênitas (DCCs) são anomalias estruturais do coração e dos grandes vasos intratorácicos presentes ao nascimento; afetam 0,8‐1 criança por 100 nascidos vivos e é o tipo mais comum de defeito congênito,1 responsável por aproximadamente um terço de todas as principais anomalias congênitas.2
De acordo com as lesões anatômicas e hemodinâmicas, as DCCs são clinicamente classificadas em diferentes subtipos dentro de um espectro de gravidade, como defeitos conotruncais, defeitos na via de saída (OFT, do inglês out flow tract), relações esquerda‐direita anormais (heterotaxia), defeitos que afetam o influxo cardíaco e cardiomiopatias.1
Aproximadamente um terço dos pacientes com DCCs tem anomalias categorizadas como graves e potencialmente letais e necessita de intervenção clínica ou cirúrgica no primeiro ano de vida, exige muitas vezes procedimentos cirúrgicos múltiplos.1 Portanto, as DCCs têm um efeito significativo sobre a morbidade, mortalidade e cuidados de saúde e apesar dos avanços nos tratamentos e nos cuidados intensivos, continuam a ser a principal causa de mortalidade infantil nos países desenvolvidos.1
À medida que os cuidados de saúde melhoram nos países mais pobres do mundo, as mortes secundárias a doenças infecciosas diminuem e as DCCs aumentam como importante causa de morbidade e mortalidade. Em 2007, as DCC foram responsáveis por 6% das mortes em crianças com menos de um ano no Brasil.3 As melhorias nas técnicas cirúrgicas e no cuidado perioperatório mudaram drasticamente a história natural das DCCs, permitiram a sobrevivência de até 95%4 dos pacientes, resultou em uma população cada vez maior de adultos que atingem a idade fértil e vivem com DCCs e as consequências das anomalias e tratamentos,5 alterações que no passado levavam à morte em idade muito jovem.
O manejo dos pacientes sobreviventes representa um novo desafio: 13,6% dos pacientes com DCCs submetidos a reparo ou tratamento paliativo apresentam malformações estruturais extracardíacas associadas,1 além do aumento do risco de arritmias, disfunção miocárdica e deficiências do neurodesenvolvimento, que são potencialmente as comorbidades com maior impacto na qualidade de vida em pacientes com DCCs: eles afetam 10% a 50% dos pacientes, de acordo com a gravidade da doença coronariana.1
A complexidade e a heterogeneidade das DCCs têm sido tradicionalmente atribuídas a etiologias multifatoriais, decorrentes de interações entre múltiplos genes e fatores ambientais.2,4 De fato, não é fácil definir com precisão a contribuição genética subjacente aos defeitos cardíacos, devido à complexidade da rede genética que controla a organogênese do coração. Entretanto, muitos estudos apontam para uma importante contribuição genética para DCCs, como maior concordância em gêmeos monozigóticos, risco de recorrência de formas relacionadas de DCCs em irmãos e a presença de formas mendelianas raras de defeitos cardíacos.1
Base do desenvolvimento do coraçãoO coração é o primeiro órgão funcional a se desenvolver em embriões de vertebrados e esse processo é estritamente controlado por uma rede de regulação gênica,2,6 que inclui fatores de transcrição, vias de sinalização, microRNAs e fatores epigenéticos.
Em mamíferos, três linhagens celulares colaboram no curso da morfogênese cardíaca: células do mesoderma cardiogênico (CMC), o proepicárdio (PE) e células cardiogênicas da crista neural (CCCN).7 O primeiro campo cardíaco (PCC) e o segundo campo cardíaco (SCC) que formam a maior proporção de miocárdio ventricular, atrial e da via de saída, além de endocárdio, sistema de condução e coxins pulmonares e aórticos, abrigam‐se no mesoderma cardiogênico.7–9 Inicialmente, o PCC forma o crescente cardíaco, que evolui para o coração tubular ou tubo cardíaco, que é o principal contribuinte para o ventrículo esquerdo inicial.
À medida que o tubo cardíaco se forma, o SCC migra para a linha média e se posiciona dorsalmente ao tubo cardíaco, compreende o aspecto dorsal‐medial da placa cardíaca, enquanto o PCC compreende o aspecto ventral. O PCC diferencia‐se como o crescente cardíaco, enquanto a diferenciação do SCC é atrasada pela sinalização inibitória de Wnt que emana da linha média.2,8 Então, ele cresce e povoa grande parte do OFT, do ventrículo direito primário e dos átrios. Fatores secretados da porção anterior do tubo cardíaco funcionam como quimioatrativos para as células do SCC, embora esses mecanismos permaneçam desconhecidos.2,8
Ambas as linhagens parecem ser controladas por sinais positivos e negativos intricados de vias como proteínas morfogenéticas ósseas (BMPs, do inglês bone morphogenetic proteins), fatores de crescimento de fibroblastos (FGF, do inglês fibroblast growth factors), vias de sinalização Sonic Hedgehog (SHH), WNT e NOTCH.2,7 O comprometimento cardiogênico inicial depende da expressão do fator de transcrição Nkx2.5 em células mesenquimais, como consequência da expressão de BMP2/4 associada a inibidores da via Wnt.9,10
As células progenitoras que surgem do PE compreendem o epicárdio e se diferenciam em fibroblastos, músculo liso dos vasos e células endoteliais das coronárias e alguns miócitos formam o septo atrioventricular (AV). A interação entre o epicárdio e o miocárdio é crucial para a maturação da câmara e o crescimento do músculo ventricular.7 Essa interação é proporcionada por uma matriz extracelular denominada geleia cardíaca, que favorece a sinalização recíproca entre o miocárdio externo e o endocárdio interno.2
Finalmente, as CCCNs originam‐se do tubo neural dorsal e migram para os arcos faríngeos 3, 4 e 6, compreendem células distais do OFT e de músculo liso da crista aorticopulmonar, bem como a inervação autonômica do coração.7–9 As CCCNs são essenciais para a maturação e septação do polo arterial do coração e contribuem para a formação do septo e da válvula.6
O coração é o primeiro órgão a romper a simetria embrionária, à medida que o tubo inicia o looping para a direita, reflete o estabelecimento global da assimetria esquerda‐direita (ED), envolve a conversa cruzada complexa entre vias como Notch, Nodal, SHH, FGF, BMP e, finalmente, restringe a sinalização Nodal ao lado esquerdo do embrião,2,6 através da atividade de células ciliares que geram um fluxo de sentido direcional de fluido extraembrionário.1,4
O estabelecimento da assimetria ED é seguido pela formação dos coxins endocárdicos dentro do OFT e do canal AV que contribui para dividir o coração nas quatro câmaras cardíacas, inicia a divisão da OFT na aorta e na via pulmonar e precede a formação das válvulas em cada extremidade do tubo cardíaco.2,4
Alterações genéticas subjacentes à DCCA maioria das DCCs ocorre como malformações isoladas, enquanto 25 a 30% delas estão associadas a anomalias extracardíacas, e alguns defeitos específicos são frequentemente encontrados em associação com síndromes genéticas conhecidas.11 Vários dados apontam que a genética contribui para a maioria dos DCCs,1 embora padrões clássicos de herança mendeliana não sejam geralmente observados.12,13
As principais anomalias cromossômicas têm sido associadas a DCCs por mais de meio século. As aneuploidias são as causas genéticas mais precocemente identificadas nas DCCs e a contribuição das anormalidades citogenéticas varia de 9% a 18%.1 O grande número de genes afetados resulta em fenótipos pleiotrópicos e graves e 98% dos fetos afetados têm pelo menos uma anormalidade extracardíaca.1
Ferramentas de investigação genética mais recentes, como o array‐CGH, foram cruciais para revelar a presença de anomalias estruturais submicroscópicas associadas a síndromes genéticas identificáveis, inclusive fenótipos de DCCs.12,14 As mutações somáticas não são uma causa comum de DCC, mas existe a possibilidade de que tenham um papel no desenvolvimento da doença em um ambiente poligênico ou multifatorial.14
Anormalidades cromossômicas e DCCsAnomalias cromossômicas clássicas detectáveis pelo cariótipo padrão normal incluem trissomia do 21 (síndrome de Down, OMIM 190685), trissomia do 13 (síndrome de Patau) trissomia do 18 (síndrome de Edwards), monossomia do X (síndrome de Turner).11 As DCCs são observadas em até 50% dos nascidos vivos com trissomia do 21, 60% a 80% dos nascidos vivos com trissomia do 13 e 33% com monossomia do X.1 Cada anormalidade cromossômica está preferencialmente associada a tipos específicos de DCC, como a que ocorre com defeitos AV e síndrome de Down ou lesões obstrutivas do ventrículo esquerdo e síndrome de Turner.1,11
Anomalias cromossômicas submicroscópicas são detectáveis por hibridização fluorescente in situ (FISH), amplificação de múltiplas sondas dependente de ligação (MLPA) e microarranjos cromossômicos (CMA). Essas técnicas aumentaram o conhecimento sobre as variações do número de cópias (CNVs): variações genômicas comuns na população que incluem deleções e duplicações com diferentes consequências genômicas. As CNVs geralmente surgem a partir de rearranjos genômicos em pontos de quebra cromossômicos comuns devido à arquitetura genômica e que não são necessariamente patológicos. Entretanto, raras CNVs podem levar a um aumento, ruptura ou redução da expressão de um ou mais elementos genômicos, levar a patologias e problemas de desenvolvimento, e são constantemente ligadas a DCCs sindrômicas e não sindrômicas.15,16
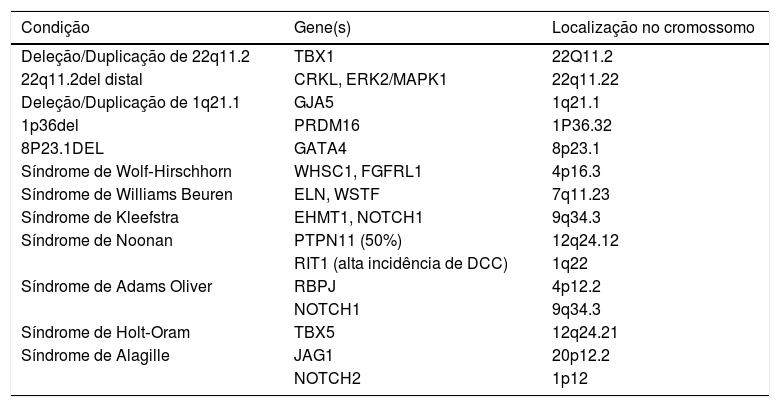
A fim de compreender a contribuição das anormalidades cromossômicas estruturais para a etiologia da doença coronariana, detalhamos a seguir os principais distúrbios genômicos (síndromes associadas à CNVs) relacionados à DCC, resumidos na tabela 1.
CNV associadas com cardiopatias congênitas.×
| Condição | Gene(s) | Localização no cromossomo |
|---|---|---|
| Deleção/Duplicação de 22q11.2 | TBX1 | 22Q11.2 |
| 22q11.2del distal | CRKL, ERK2/MAPK1 | 22q11.22 |
| Deleção/Duplicação de 1q21.1 | GJA5 | 1q21.1 |
| 1p36del | PRDM16 | 1P36.32 |
| 8P23.1DEL | GATA4 | 8p23.1 |
| Síndrome de Wolf‐Hirschhorn | WHSC1, FGFRL1 | 4p16.3 |
| Síndrome de Williams Beuren | ELN, WSTF | 7q11.23 |
| Síndrome de Kleefstra | EHMT1, NOTCH1 | 9q34.3 |
| Síndrome de Noonan | PTPN11 (50%) | 12q24.12 |
| RIT1 (alta incidência de DCC) | 1q22 | |
| Síndrome de Adams Oliver | RBPJ | 4p12.2 |
| NOTCH1 | 9q34.3 | |
| Síndrome de Holt‐Oram | TBX5 | 12q24.21 |
| Síndrome de Alagille | JAG1 | 20p12.2 |
| NOTCH2 | 1p12 |
A microdeleção humana mais comum varia de 0,7 a 3 Mpb, que afeta cerca de uma em 4.000 pessoas, resulta em um amplo espectro de fenótipos característicos da síndrome de DiGeorge (OMIM 188400), síndrome velocardiofacial (OMIM 192430) e síndrome de Shprintzen (OMIM 182212), abrange DCC, especialmente defeitos conotruncais, anormalidades do palato, hipocalcemia, imunodeficiência, características faciais distintas e anormalidades do desenvolvimento neurológico.1
A síndrome da deleção de 22q11.2 (22q11del) inclui alterações no fator de transcrição de T‐Box TBX1, o que evidencia a importância da regulação da transcrição de SHF, uma vez que o geneTBX1 é peça central no desenvolvimento adequado da OFT do miocárdio4,17 e é expressa apenas na SHF, não na FHF nem nas CCCN. O TBX1 também regula a expressão de fatores de crescimento, como o fator de crescimento de fibroblastos 8 (FGF8), que ativa a diferenciação das CCCN. Portanto, os defeitos da OFT, tipicamente truncus arteriosus, tetralogia de Fallot e anormalidades do arco aórtico estão altamente associados à 22q11del, bem como defeitos craniofaciais, inclusive fenda palatina,18 hipoplasia do timo e da paratireoide.
A maioria dos pacientes com 22q11.2del tem deleções de novo, resulta de recombinação não homóloga entre repetições de cópia baixas que flanqueiam a região crítica do cromossomo. Deleções mais proximais mantêm um fenótipo próximo à síndrome de DiGeorge, inclusive imunossupressão, enquanto deleções mais distais apresentam características clínicas incompletas: genes que interagem funcionalmente com TBX1, mas localizados distalmente a ele, como CRKL, ERK2/MAPK1, têm sido propostos como a etiologia de DCC em deleções mais distantes, particularmente a haploinsuficiência do gene MAPK1.11
Síndrome da duplicação de 22q11.2 (OMIM 608363)Fenotipicamente semelhante à microdeleção correspondente, é difícil estabelecer qualquer correlação clara genótipo‐fenótipo para a microduplicação de 22q11.2; entretanto, os defeitos parecem pertencer a diferentes vias patogênicas: defeitos septais e lesão obstrutiva do ventrículo esquerdo, geralmente associada a distúrbios neurológicos e retardo de crescimento. A prevalência de DCC na duplicação de 22q11.2 é menor em comparação com a deleção da mesma região, mas a base molecular também considera o gene TBX1 como candidato, pois é superexpresso e pode interagir com outros genes dentro e fora da região cromossômica afetada.11 A patogenicidade da microduplicação ainda é difícil de definir, pois a maioria dos portadores parentais tem um fenótipo normal. Mas o notório enriquecimento dessa microduplicação em pacientes com deficiências do desenvolvimento neurológico e a maior ocorrência de uma segunda CNV em portadores afetados sugerem que as microduplicações de 22q11distais podem atuar como um locus de susceptibilidade para a deficiência do desenvolvimento neurológico.19
Deleção e duplicação de 1q21.1A DCC é uma característica importante da síndrome de deleção do 1q21.1 (OMIM 612474), com um fenótipo heterogêneo, inclusive incapacidade intelectual leve a moderada, microcefalia e DCC, como obstruções no lado esquerdo (40%), defeitos septais (27%) e defeitos conotruncais (20%).11 Não houve diferenças fenotípicas significantes entre portadores de deleções com diferentes pontos de ruptura.20 A síndrome dw duplicação de 1q21.1 (OMIM 612475) é muito menos comum e inclui o GJA5, descrito como um gene de suscetibilidade para DCC, notadamente a tetralogia de Fallot, e foi descrito como tendo sofrido mutação em pacientes com DCC não sindrômica.4,18,21 O gene GJA5, que codifica a conexina Cx40 (Cx40),22 é uma proteína da junção do gap cardíaco – proteína do canal da membrana celular que interconecta o citoplasma das células vizinhas e é responsável pela condução célula a célula do potencial de ação. A conexina Cx40 é ricamente expressa no miocárdio atrial e no sistema de condução atrioventricular.4 O desequilíbrio na expressão dessa conexina está associado com maior propensão a arritmias. Além disso, pacientes com formas mutadas também apresentam atraso no desenvolvimento e características dismórficas.21,22
Síndrome da deleção de 1p36A síndrome de 1p36Del (OMIM 607872) é o segundo distúrbio de microdeleção mais comum e é caracterizada por deficiência intelectual, epilepsia, características dismórficas, distúrbios metabólicos e neuromusculares. Deleções terminais e intersticiais são observadas com pontos de quebra altamente variáveis. A DCC está presente em 50% dos casos, principalmente cardiomiopatia e alta prevalência de não compactação do ventrículo esquerdo.23 O gene que codifica o fator de transcrição PRDM16 localiza‐se dentro da região crítica da síndrome 1p36 e está ligado à não compactação do ventrículo esquerdo não sindrômica.23,24
Deleção de 8p23.1Deleções que envolvem o cromossomo 8p23.1 variam de grandes deleções que incluem o telômero 8p e detectáveis por cariotipagem de rotina a pequenas deleções intersticiais que resultam em diferentes fenótipos, particularmente hérnia diafragmática e DCC.4,24 Defeitos cardíacos são observados em 94% dos casos, variam de defeitos septais isolados a DCC mais complexas, como a tetralogia de Fallot e a síndrome do coração esquerdo hipoplásico.4
A alta incidência de DCC é devida principalmente à ausência ou à expressão desequilibrada do fator de transcrição GATA4, que é conhecido por ter um papel importante no desenvolvimento do coração em humanos. A haploinsuficiência do gene GATA4 tem sido descrita como a etiologia da DCC não sindrômica em modelos animais e em famílias, especialmente defeitos septais.4,11,15 Pacientes com deleções de 8p23.1 podem ter doença coronariana mais grave e complexa quando comparados com pacientes com mutações no gene GATA4isoladas, sugere que outros genes localizados na região podem ter um papel no fenótipo da DCC.25 Entre esses genes, a haploinsuficiência do gene do fator de transcrição SOX7 é um dos mais prováveis de exacerbar os efeitos da deleção do GATA4.25
Síndrome de Wolf‐Hirschhorn (Deleção 4pter)A síndrome de Wolf‐Hirschhorn (WHS, OMIM 194190) é causada pela perda da porção distal da região 4p, com o ponto de ruptura geralmente entre 4p15 e 4p16.26 A frequência estimada é em torno de 1:50.000 nascidos vivos.26 O fenótipo inclui características faciais distintas (conhecidas como fácies de “capacete grego”, com nariz distinto, hipertelorismo ocular, filtro labial curto, testa alta, sobrancelhas arqueadas), atraso neurológico e de crescimento e convulsões. A DCC é descrita em 50% dos casos, particularmente defeitos septais leves e persistência do ductus arteriosus, embora tenham sido relatados defeitos cardíacos mais graves.26,27 O gene mais provável implicado no fenótipo de DCC é oWHSC1,uma histona metiltransferase de lisina, que interage com o fator de transcrição cardíaca Nkx2.5 especialmente durante a formação de septo cardíaco.4 Outro candidato é o gene FGFRL1, que codifica um membro da família de receptores de crescimento de fibroblastos expresso no cérebro, placódios cranianos, arcos faríngeos e coração.
Síndrome de Williams‐BeurenA síndrome de Williams‐Beuren (WBS, OMIM 194050) é causada por uma deleção típica de 1,5‐1,8 Mbp na região 7q11.23, envolve cerca de 28 genes, afeta 1:7500 a 1:10.000 indivíduos. A maioria dos pacientes é heterozigótica para uma deleção de 1,5‐1,8 Mbp.4 Anomalias cardiovasculares estão presentes em 75% dos indivíduos, geralmente estenose aórtica supravalvar e estenose pulmonar, o que pode ser explicado pela haploinsuficiência do gene da elastina (ELN), causa deficiência ou deposição anormal de elastina na parede arterial, leva à proliferação de células de músculo liso arterial e subsequente hiperplasia intimal.28 Mutações de ponto no gene ELN foram relatadas em pacientes com estenose aórtica supravalvar não sindrômica.4
Outras DCCs, como defeitos septais e tetralogia de Fallot, são descritas em 6‐10% dos pacientes e não podem ser explicadas pela deleção do gene ELN. Modelos animais indicam que a deleção de outro gene na região 7q11.23 ‐ BAZ1B, também conhecido como fator de transcrição da síndrome de Williams (WSTF), pode ser responsável por esses defeitos.4,29 O gene WSTF codifica uma subunidade em três complexos de remodelação da cromatina dependentes de ATP que é crucial para as cascatas de transcrição gênicas normais no coração em desenvolvimento.29
Síndrome de KleefstraA síndrome de Kleefstra (KLEFS1, OMIM 610253) é causada pela microdeleção da região 9q34.3 ou, menos comum, por mutações de ponto no gene histona‐lisina N‐metiltransferase 1 eucromática (EHMT1).A síndrome de Kleefstra é uma doença clínica reconhecível com características típicas da face (face plana com hipertelorismo ocular, sinofris, lábio inferior evertido, macroglossia e narinas antevertidas) e DCC em aproximadamente 40% dos pacientes,4,30 inclusive defeitos septais, coarctação de aorta, estenose pulmonar ou tetralogia de Fallot.16 Um importante atraso do desenvolvimento, alterações geniturinárias, constipação crônica e epilepsia também são descritos.30
Mutação de gene único e DCCO sequenciamento de próxima geração (NGS, do inglês next generation sequencing) abriu as portas para o entendimento da genética de doenças complexas, como as DCCs, além de grandes variações estruturais, permitiu a identificação de mutações que, de outra forma, seriam indetectáveis.1
A rede de controle do desenvolvimento cardíaco é vasta e intricada e mutações genéticas, inclusive aquelas com ganho de função e perda de função, que afetam esse complexo processo desempenham um papel significativo na genética das DCCs.2
Genes mutados em DCC são geralmente agrupados de acordo com a função e o envolvimento em vias específicas, uma vez que isso esclarece a compreensão desses genes na formação cardíaca.
Abaixo mencionamos algumas das vias/mecanismos importantes e síndromes associadas relacionadas à DCC.
Síndrome de Noonan e RASopatiasAs RASopatias são um grupo de síndromes causadas por mutações em genes da via Ras‐MAPK, que é essencial para o ciclo celular, com papéis reguladores na proliferação, diferenciação, crescimento e metabolismo celular. Portanto, sua desregulação nessa cascata é responsável por profundas consequências no desenvolvimento.11 Elas incluem a síndrome de Noonan (NS) e outras doenças relacionadas à síndrome de Noonan (DRSN), inclusive a síndrome cardio‐facio‐cutânea (CFC; OMIM 115150), síndrome de Costello (SC; OMIM 218040) e SN com lentigos múltiplos (SNLM; também conhecida como síndrome LEOPARD; OMIM 151100). Esses são distúrbios do desenvolvimento que se sobrepõem clinicamente e que compartilham muitos traços característicos, como dismorfismo facial, baixa estatura e anormalidades cardíacas, com fenótipos que são amplos e heterogêneos, e o diagnóstico diferencial entre eles pode ser difícil.31
A síndrome de Noonan é uma das síndromes genéticas mais comuns associadas à DCC, com uma prevalência estimada de 1:1000 a 1:2500 nascidos vivos. É um distúrbio clinicamente heterogêneo, transmitido como um traço autossômico dominante.13 As características clínicas incluem baixa estatura, características dismórficas (como face triangular, hipertelorismo, baixa implantação das orelhas e ptose) defeitos linfáticos, hematológicos, esqueléticos e ectodérmicos. Além disso, os pacientes podem apresentar comprometimento neurológico variável, variam de incapacidade intelectual moderada a capacidade superior; no entanto, crianças com doença cardíaca grave tendem a apresentar capacidade cognitiva mais baixa.30 Perda auditiva, falta de coordenação, distúrbios do humor, hiperatividade e déficit de atenção também foram descritos.11,13,32
DCC ocorre em 60‐90% dos pacientes com RASopatias, com menor ocorrência em SNLM. As anormalidades cardíacas mais frequentes são estenose da válvula pulmonar, defeito do septo atrioventricular (DSAV), defeitos septais e cardiomiopatia hipertrófica.8,11,31
Diferentes genes foram identificados como responsáveis pelo fenótipo da síndrome de Noonan ou condições correlacionadas. Mutações missense no gene PTPN11– localizado na região 12q24.1 – são responsáveis por aproximadamente 50% dos casos. Outros 12 genes estão envolvidos e junto com o PTPN11 respondem por aproximadamente 90% dos casos afetados: KRAS, SOS1, RAF1, NRAS, BRAF, SHOC2, PPP1CB, CBL, RRAS, RIT1, LZTR1eSOS2. A maioria desses genes codifica proteínas que estão envolvidas na via de sinalização da proteína quinase ativada por mitógeno RAS (MAPK). A via RAS/MAPK é uma importante cascata de sinalização que permite às células responder adequadamente a múltiplos estímulos extracelulares, inclusive fatores de crescimento, hormônios e citocinas, controla praticamente todos os processos celulares. A maioria dessas mutações leva a um aumento da transdução de sinal ao longo dessa via, causa ativação contínua da MAPK durante o desenvolvimento.13,33SOS1, RIT1, eRAF1são os genes que mais frequentemente sofrem mutações e a prevalência de DCC em pacientes com mutações no gene RIT1 é particularmente alta (90%).11,13
Defeitos do septo atrioventricular (DSAV) são encontrados especialmente em pacientes com mutações nos genes PTPN11 e RAF1 e DSAV parcial associada a obstruções do lado esquerdo, estenose valvar pulmonar ou cardiomiopatia hipertrófica devem ser consideradas como marcadores para Noonan ou síndromes correlacionadas.11
Síndrome de Adams‐OliverA síndrome de Adams‐Oliver (AOS1, OMIM 100300) é um distúrbio de desenvolvimento raro, caracterizado por aplasia cutânea congênita do vértice do couro cabeludo, defeito transverso terminal dos membros, com alta variabilidade intra‐ e interfamiliar.11,34 As malformações cardiovasculares ocorrem em 13% a 20% dos pacientes e diferentes aspectos anatômicos tipos têm sido relatados: lesões obstrutivas do lado esquerdo, defeitos septais e conotruncais e atresia tricúspide. As lesões do lado esquerdo são predominantes e ocorrem em múltiplos níveis.34
Os genes clássicos envolvidos na síndrome de Adams‐Oliver incluem ARHGAP31, DOCK6, RBPJ e EOGT e apenas 9% dos pacientes com essas mutações têm DCC, particularmente, defeitos do septo.34
O gene RBPJ foi proposto como candidato à síndrome de Adams‐Oliver com DCC mais complexa, pois codifica uma proteína altamente preservada que coordena a ativação transcricional dos genes‐alvo da via NOTCH, é uma chave importante para a formação de células mesenquimais, esqueléticas, vasculares e formação da epiderme e folículos pilosos.35 Demonstrou‐se que as variantes do gene NOTCH1, pertencentes à via de sinalização NOTCH, estão relacionadas à síndrome de Adams‐Oliver com DCC e foi proposto que os defeitos nos membros e no couro cabeludo são secundários à vasculopatia causada pela haploinsuficiência do NOTCH1.11 O gene NOTCH1 não foi apenas implicado na DCC não sindrômica, mas também é conhecido por ser essencial para a transformação da epiderme em células mesenquimais migratórias, definição do território valvar, além de ser amplamente expresso na OFT.35
Síndrome de Holt‐OramA síndrome de Holt‐Oram (SHO, OMIM 142900) afeta 1:100.000 indivíduos e pode ser esporádica ou hereditária, transmitida como doença autossômica dominante causada por mutações non sense ou frameshift no gene TBX5,13,16 ou até mesmo duplicações que envolvem esse gene na região 12q.35 TBX5 é um fator de transcrição que é um conhecido regulador‐chave da organogênese do coração, especialmente quando em combinação com outros fatores de transcrição, como NKX2.5 e GATA4.2,13,17
Esta síndrome é caracterizada por malformações dos membros superiores e DCC, especialmente defeitos do septo e distúrbios de condução. As anormalidades dos membros superiores estão sempre presentes, envolvem estruturas derivadas do raio radial e são mais comumente bilaterais e assimétricas, variam de achados radiológicos subclínicos a focomelia. Os defeitos cardíacos são tipicamente defeitos septais, mas doenças cardíacas mais complexas já foram descritas. Anormalidades da condução cardíaca também são usualmente encontradas. Nenhuma correlação pode ser feita entre a gravidade das malformações cardíacas e nos membros.36
Síndrome de AlagilleA síndrome de Alagille (ALGS1, OMIM 118450) é um distúrbio multissistêmico com prevalência estimada de 1:70.000 recém‐nascidos, que afeta o coração, fígado, olhos, face e esqueleto, e a DCC está presente em 90% dos casos. O envolvimento da via de saída pulmonar é o tipo mais comum de DCC descrita, entre os quais a estenose pulmonar valvar e/ou arterial e a tetralogia de Fallot são usualmente mencionadas. A escassez de ductos biliares interlobulares e a consequente colestase também são uma característica clínica importante.13
A grande maioria dos pacientes com síndrome de Alagille (> 90%) tem mutações no gene JAG1, que codifica uma ligante sinalizadora de NOTCH. Casos selecionados (< 1%) têm mutações no gene NOTCH2.37 O gene JAG1 está fortemente correlacionado com malformações cardiovasculares e é subjacente à DCC não sindrômica, mais frequentemente a tetralogia de Fallot.1
Como as DCCs podem ser uma manifestação isolada da síndrome de Alagille, pacientes com histórico familiar de tetralogia de Fallot ou aqueles com estenose ou hipoplasia de ramo de artéria pulmonar devem ter direito a testes genéticos, mesmo que outras características fenotípicas estejam ausentes.1,13
Genes envolvidos no controle epigenéticoNovas abordagens têm sido usadas na busca pela compreensão da etiologia e variabilidade fenotípica de doenças genéticas complexas. O estudo da epigenética – alterações genômicas que não envolvem modificações na sequência de DNA – sugere que a estrutura da cromatina e/ou os distúrbios epigenéticos podem levar a alterações na transcrição de múltiplos genes e vias metabólicas que podem desempenhar um papel fundamental nas DCC. Diversos estudos têm demonstrado a importância de vários mecanismos de regulação epigenética durante a cardiogênese.38
Diversos estudos demonstram a importância de vários mecanismos de regulação epigenética durante a cardiogênese. Alterações da metilação do DNA, especialmente nas ilhas CpG próximas a fatores de transcrição, foram identificadas em pacientes com malformações cardíacas.38 Estudos com sequenciamento de próxima geração identificaram enriquecimento significativo para mutações em genes que envolvem modificação de histonas em pacientes com DCC, especialmente H3K4, H2K7, H3K9 e H3K27, sugere que a modificação de histonas pode ser significativa na patologia da doença isolada.1,4
A investigação do remodelamento da cromatina em organismos modelo mostrou que a modificação dinâmica da estrutura da cromatina desempenha um papel importante na regulação da expressão gênica durante o desenvolvimento do coração.13 Mutações de novo que afetam os genes de regulação da cromatina contribuem para cerca de 3% das DCCs. Além disso, os genes reguladores da cromatina incluem cerca de 600 genes que regulam a expressão gênica dinâmica e alteram fatores epigenéticos ou catalisam alterações na estrutura da cromatina.1 Portanto, genes que codificam proteínas que modificam ou se ligam a histonas têm sido implicados como a etiologia de síndromes que causam DCCs, como:
Síndrome de KabukiA síndrome de Kabuki (KABUK1, OMIM 147920) é um distúrbio genético que afeta 1:30.000 recém‐nascidos e causa atraso no desenvolvimento, dismorfismo facial e lesões obstrutivas do lado esquerdo, que inicialmente levantaram uma suspeita de envolvimento do cromossomo X, embora defeitos do septo e conotruncais também possam ser detectados.11 De fato, apesar da identificação do gene MLL2 – uma histona metiltransferase – como causa primária da síndrome de Kabuki através do sequenciamento do genoma completo, deleção de novo parcial ou completa de genes do cromossomo X, que codifica as modificadoras de histonas KMT2D e KDM6A que interagem com o gene MLL2 também podem resultar em um fenótipo da síndrome de Kabuki.11,13
Síndrome ChargeA síndrome Charge (OMIM 214800) é um acrônimo que significa coloboma, cardiopatia, atresia coanal, atraso do crescimento e desenvolvimento, hipoplasia dos genitais, anomalias dos pavilhões auriculares/surdez (do inglês iris Coloboma, Heart malformation, choanal Atresia, Retarded growth and development, Genital hypoplasia and Ear anomalies and deafness), embora outras malformações e alterações comportamentais possam estar presentes e os critérios diagnósticos tenham sido refinados várias vezes.39 Ela afeta 1:8.000 a 1:10.000 recém‐nascidos, cerca de 70% dos pacientes têm DCC e cerca de metade deles apresenta defeitos conotruncais maiores, como a tetralogia de Fallot e dupla via de saída do ventrículo direito, embora outros defeitos da OFT, como a síndrome do coração esquerdo hipoplásico, também sejam descritos.13,39 Mais de dois terços dos casos são causados por mutações non sense ou frame shift no gene CHD7, que codifica uma proteína modificadora da cromatina, embora alterações no gene da semaforina (SMA3E) possam resultar em fenótipo semelhante.40 A proteína CHD7 é essencial para a migração da crista neural, o que pode explicar a alta frequência de defeitos da OFT.13,39
Síndrome de Koolen‐De VriesA Síndrome de Koolen‐De Vries (KDVS, OMIM 610443) é causada pela deleção do locus 17q21.31 ou mutação do gene KANSL1, localizado no loco supracitado, caracteriza‐se por grave déficit intelectual, hipotonia, convulsões e dismorfismo facial. A DCC está presente em 27% dos casos, com defeitos marcadamente septais, embora a estenose pulmonar também possa ser descrita.1,41 Estudos recentes identificaram que o gene KANSL1 desempenha um papel como gene modificador em pacientes com 22q11.2DS.42
DCC não sindrômicaA grande maioria das DCCs – cerca de 70% – ocorre como malformações isoladas,11,12,43 inclusive as mais complexas: atresia da tricúspide, transposição das grandes artérias, síndrome do coração esquerdo hipoplásico e atresia pulmonar. Vários novos genes com herança mendeliana foram identificados e estudos de famílias afetadas não só lançaram luz sobre os padrões de herança, mas também têm sido essenciais para a compreensão da complexa organogênese do coração, uma vez que os genes etiologicamente ligados às DCCs afetam diretamente o desenvolvimento embriológico e também podem desempenhar um papel na regulação do coração durante toda a vida.12 A tecnologia de sequenciamento de próxima geração (NGS) abriu as portas para se descobrir a importância das variantes de novo sem herança mendeliana clara, variantes com penetrância reduzida e alterações somáticas, entre outras.12,13
A maioria das mutações identificadas é família‐específica e não pode ser considerada uma causa comum de DCC, mas é possível que múltiplas variantes possam ter um papel no desenvolvimento da doença como ambiente poligênico, embora a interpretação dessas variantes possa ser muito desafiadora e não é sempre é possível estabelecer sua patogenicidade. Essas associações podem ser altamente significativas do ponto de vista estatístico e de pesquisa, mas têm relevância clínica baixa.14
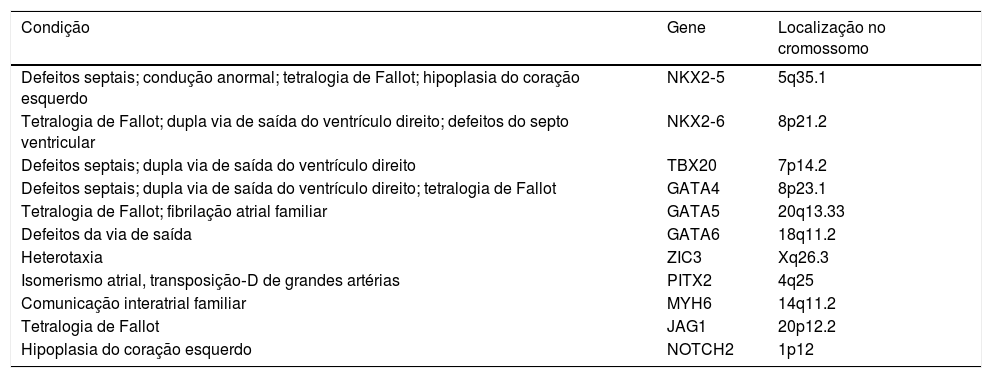
Em muitas famílias e indivíduos com DCC, as variações nos genes expressos durante a formação do coração estão presentes com diferentes perfis de herança, sugere um continuum entre formas mendelianas e complexas das doenças, além de distúrbios de gene único, como exemplificado abaixo e listados na tabela 2.14
Genes associados a cardiopatias congênitas não sindrômicas
| Condição | Gene | Localização no cromossomo |
|---|---|---|
| Defeitos septais; condução anormal; tetralogia de Fallot; hipoplasia do coração esquerdo | NKX2‐5 | 5q35.1 |
| Tetralogia de Fallot; dupla via de saída do ventrículo direito; defeitos do septo ventricular | NKX2‐6 | 8p21.2 |
| Defeitos septais; dupla via de saída do ventrículo direito | TBX20 | 7p14.2 |
| Defeitos septais; dupla via de saída do ventrículo direito; tetralogia de Fallot | GATA4 | 8p23.1 |
| Tetralogia de Fallot; fibrilação atrial familiar | GATA5 | 20q13.33 |
| Defeitos da via de saída | GATA6 | 18q11.2 |
| Heterotaxia | ZIC3 | Xq26.3 |
| Isomerismo atrial, transposição‐D de grandes artérias | PITX2 | 4q25 |
| Comunicação interatrial familiar | MYH6 | 14q11.2 |
| Tetralogia de Fallot | JAG1 | 20p12.2 |
| Hipoplasia do coração esquerdo | NOTCH2 | 1p12 |
A família NK2 é constituída por genes homeobox que desempenham papéis cruciais no desenvolvimento do coração, regulam processos essenciais, como a expressão gênica espacial e temporal.8,9,44 O gene NKX2‐5é expresso em ambos, o primeiro e o segundo campos cardíacos, como um dos primeiros marcadores da diferenciação cardiomiogênica e é fundamental para a hierarquia reguladora cardíaca.2 Várias mutações foram descritas, levaram principalmente a defeitos septais e anormalidades da condução atrioventricular,2 mas também foram descritas DCC mais complexas, como a tetralogia de Fallot e a hipoplasia do coração esquerdo.45 Estudos recentes têm se concentrado na região reguladora do gene NKX2‐5, propõem que essas variantes não codificantes podem melhorar a transcrição e alterar a rede que controla a morfogênese cardíaca. Também foi postulado que essas versões mutantes podem se ligar a promotores de genes não específicos e permitir que os cofatores induzam um efeito mais forte do que o usual, o que pode explicar as grandes variações de fenótipos em indivíduos afetados.45
O gene NKX2‐6 se sobrepõe parcialmente ao NKX2‐5 nos perfis de expressão temporal e espacial e nas características funcionais durante a embriogênese. As mutações que causam perda de função do NKX2‐6 já foram identificadas em pacientes com tetralogia de Fallot, dupla via de saída do ventrículo direito e defeitos do septo ventricular.2
Mutações na família TBXA família do fator de transcrição toolbox (TBX) é um grupo de seis proteínas que compartilham um domínio de ligação de DNA altamente conservado e com papel significativo no desenvolvimento de células progenitoras cardíacas – especialmente no segundo campo cardíaco – bem como na padronização das câmaras e OFT.2,17
O gene TBX1 é expresso no mesênquima e no epitélio da faringe e é um dos principais determinantes genéticos de distúrbios cardíacos e craniofaciais, é incluído no conjunto de genes deletados na síndrome de 22q11del. Mutações no gene TBX5 foram associadas à síndrome de Holt‐Oram, como descrito. Existem muito poucos casos de doença coronariana isolada relacionados a mutações nesses dois genes.44
Mutações no gene TBX20, por outro lado, têm sido associadas com defeitos do septo atrial e ventricular e valvulogênese aberrante: o TBX20 é necessário nas linhagens endoteliais para septação, regulagem do versican, um proteoglicano da matriz extracelular e a proliferação e diferenciação dos cardiomiócitos nos septos.4,46 Mutações no TBX20 aumentam a suscetibilidade da dupla via de saída do ventrículo direito em humanos e também têm sido associadas de maneira causal à cardiomiopatia dilatada.2
Mutações na família GATAA família dos fatores de transcrição de “dedos de zinco” GATA compreende seis membros: GATA 1 a GATA 6, que se ligam à sequência de bases (A/T) GATA (A/G) na região reguladora de vários genes. A maioria dos tecidos de origem mesodérmica ou endodérmica expressa pelo menos um dos seguintes: GATA4, GATA5 ou GATA6 e todos os três estão presentes no mesoderma pré‐cardíaco.2,7 Experimentos em modelos animais mostraram que o silenciamento dos genes GATA pode resultar em DCC, varia de defeitos valvulares‐septais a acardia.44
GATA4 é o membro mais investigado e também um dos primeiros fatores de transcrição expressos no desenvolvimento de células cardíacas.17,47 Uma diminuição na expressão do gene GATA4 leva a várias formas de DCC, como defeitos do septo atrioventricular, dupla via de saída do ventrículo direito e formas familiares da tetralogia de Fallot.2,17,47
O gene GATA5 pode promover o destino dos cardiomiócitos a partir de células‐tronco embrionárias murinas.6 Pouco se sabe sobre as mutações do GATA5 em humanos, mas três mutações em heterozigose foram identificadas em famílias com malformações anatômicas cardíacas ou fibrilação atrial familiar2,47 e na tetralogia de Fallot esporádica.2
O gene GATA6 é altamente expresso não apenas no coração em desenvolvimento – mesoderme pré‐cardíaca, tubo cardíaco – mas também em cardiomiócitos adultos em ventrículos e átrios humanos e células de músculo lisovascular.2,47 A deleção do GATA6 em células de músculo liso derivadas da crista neural pode resultar em defeitos na OFT, como arco aórtico interrompido e persistência do truncus arteriosus, fenótipos associados à expressão gravemente diminuída do SEMA3C.17,47 A formação do coxim endocárdico também é afetada pelo GATA6, portanto mutações nesse gene têm sido implicadas na tetralogia de Fallot não sindrômica e no defeito do septo atrioventricular.2
Mutações no gene ZIC3O gene ZIC3 codifica um fator de transcrição do “dedo de zinco” que está envolvido no desenvolvimento do eixo esquerdo‐direito (ED), conhecido como gene da heterotaxia. Localizado no cromossomo X, as mutações que causam a perda de função em Zinc3 levam à heterotaxia ligada ao X e DCC isolada,2,14 como a transposição‐D das grandes artérias e a dupla via de saída do ventrículo direito.48
Mutações no gene PITX2O gene PITX2 pertence à família homeobox de fatores de transcrição da pituitária que desempenha um papel tanto na ligação do DNA quanto do RNA e consiste em três isoformas: PITX2a, PITX2b e PITX2c. A assimetria esquerda‐direita do coração depende da expressão de PITX2 na via nodal do lado esquerdo, com ativação de vias de ciclo celular wnt‐dependente downstream e sua repressão à direita.2,49 A perda de função do PITX2 de qualquer isoforma causa isomerismo atrial grave, ventrículo esquerdo com dupla via de entrada, transposição‐D de grandes artérias e persistência do truncus arteriosus.49
Genes que codificam componentes do sarcômero cardíacoGenes sarcoméricos são amplamente reconhecidos como candidatos a diversas cardiomiopatias familiares, mas alguns genes também têm sido associados a doenças cardíacas estruturais.4,50 Mutações no gene MYH6 (cadeia pesada da miosina 6) foram identificadas em formas familiares de comunicação interatrial (CIA) e a regulação molecular envolve fatores de transcrição como GATA4 e TBX5. A incidência de CIA também pode estar super‐representada na não compactação do ventrículo esquerdo, causada pela mutação do geneMYH7,4 que também está relacionada à anomalia de Ebstein.50,51
Genes da via de NotchA sinalização de Notch é uma via altamente conservada que faz a mediação da comunicação intercelular local e regula o padrão celular e é crucial em órgãos com arquitetura complexa.34 A via Notch é particularmente importante durante a formação e morfogênese do canal atrioventricular e da OFT e mutações dos genes envolvidos em seres humanos resultam em deficiências e síndromes de desenvolvimento cardiovascular muito específicas, como a de Alagille ou Adams‐Oliver.1,34 Mutações no gene JAG1 podem estar implicadas em casos isolados de DCC, especialmente na tetralogia de Fallot.1 As mutações do NOTCH1 foram associadas, dentro de uma única família, com uma variação de DCC de válvula aórtica bivalvular à síndrome do coração esquerdo hipoplásico. O geneGALNT11 foi associado à heterotaxia em seres humanos.1
Genes ciliaresOs genes ciliares têm múltiplas funções, inclusive sinalização, propulsão de fluido extracelular e controle do ciclo celular, e mutações nesses genes podem causar diversos distúrbios humanos com fenótipos pleiotrópicos. No desenvolvimento do coração, o papel mais bem compreendido para os cílios é o estabelecimento da assimetria esquerda‐direita, portanto mutações que afetam a motilidade ciliar podem resultar em heterotaxia e DCC.1 Em modelos animais, mutações em genes que codificam componentes do complexo motor de dineína (Dnah11/LRD e Dnah5) resultam em anormalidades DE cardíacas e viscerais.1 Não surpreendentemente, 12,1% dos pacientes com discinesia ciliar primária apresentam algum tipo de defeito de lateralidade, com ou sem defeitos cardíacos.52
ConclusãoO desenvolvimento do coração é extremamente complexo e exige interações entre inúmeros fatores moleculares e epigenéticos. À medida que o cuidado do paciente com DCC evolui e permite que ele cresça e se reproduza, a compreensão do papel genético, particularmente na DCC esporádica, aumenta. No tratamento à beira do leito, o reconhecimento das alterações genéticas subjacentes à cardiopatia pode ser útil na definição do prognóstico e na antecipação de complicações, como resposta inflamatória sistêmica, arritmias e insuficiência cardíaca precoce.
Com a tecnologia de sequenciamento de próxima geração, nossa compreensão da biologia da DCC se expandiu rapidamente, mas ainda há muitas questões a serem respondidas, pois os fundamentos genéticos de mais de 50% dos casos permanecem desconhecidos. A extrema heterogeneidade genética e clínica e a fraca correlação genótipo‐fenótipo tornam esse caminho ainda mais desafiador.
FinanciamentoMinistério da Ciência e Tecnologia/Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCT/CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), Fundação de Amparo à Pesquisa do Distrito Federal (FAP‐DF).
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Saliba A, Figueiredo AC, Baroneza JE, Afiune JY, Pic‐Taylor A, Oliveira SF, et al. Genetic and genomics in congenital heart disease: a clinical review. J Pediatr (Rio J). 2020;96:279–88.




