

Discuss evidence referring to the genetic role in congenital heart diseases, whether chromosomic alterations or monogenic diseases.
Data sourceLILACS, PubMed, MEDLINE, SciELO, Google Scholar, and references of the articles found. Review articles, case reports, book chapters, master's theses, and doctoral dissertations were included.
Summary of findingsCongenital heart diseases are among the most common type of birth defects, afflicting up to 1% of the liveborn. Traditionally, the etiology was defined as a multifactorial model, with both genetic and external contribution, and the genetic role was less recognized. Recently, however, as the natural evolution and epidemiology of congenital heart diseases change, the identification of genetic factors has an expanding significance in the clinical and surgical management of syndromic or non-syndromic heart defects, providing tools for the understanding of heart development.
ConclusionsConcrete knowledge of congenital heart disease etiology and recognition of the genetic alterations may be helpful in the bedside management, defining prognosis and anticipating complications.
Discutir as evidências referentes ao papel genético em cardiopatias congênitas, sejam alterações cromossômicas ou doenças monogênicas.
Fonte de dadosLilacs, PubMed, Medline, SciELO, Google Scholar e referências dos artigos encontrados. Artigos de revisão, relatos de casos, capítulos de livros, dissertações de mestrado e teses de doutorado foram incluídos.
Síntese dos dadosAs cardiopatias congênitas estão entre os tipos mais comuns de defeitos congênitos, afetando até 1% dos nascidos vivos. Tradicionalmente, a etiologia era definida como um modelo multifatorial, com contribuição tanto genética quanto externa, sendo o papel genético menos reconhecido. Recentemente, no entanto, à medida que a evolução natural e a epidemiologia das cardiopatias congênitas mudaram, a identificação de fatores genéticos tem adquirido importância crescente no tratamento clínico e cirúrgico de defeitos cardíacos sindrômicos e não-sindrômicos, fornecendo ferramentas para a compreensão do desenvolvimento do coração.
ConclusõesO conhecimento concreto da etiologia das cardiopatias congênitas e o reconhecimento das alterações genéticas podem ser úteis no tratamento à beira do leito, definindo o prognóstico e antecipando as complicações.
Congenital heart diseases (CHDs) are structural anomalies of the heart and endothoracic great vessels present at birth; they afflict 0.8–1 child per 100 live births and are the most common type of congenital defect,1 accounting for approximately one-third of all major congenital anomalies.2
According to anatomic and hemodynamic lesions, CHDs are clinically classified into different subtypes within a spectrum of severity, such as conotruncal defects, outflow tract (OFT) defects, abnormal left-right (LR) relationships (heterotaxy), defects affecting the inflow, and cardiomyopathies.1 Approximately one-third of the patients with CHD have anomalies categorized as severe and potentially lethal and require clinical or surgical intervention in the first year of life, often demanding multiple surgical procedures.1 Therefore, CHD has a significant effect on morbidity, mortality, and health care costs, and despite progress in treatments and intensive care, it remains the major cause of child mortality in developed countries.1
As health care improves in the world's poorest countries, deaths secondary to infectious diseases decrease and CHD rises as an important cause of morbidity and mortality. In 2007, CHDs were responsible for 6% of deaths in children <1 year old in Brazil.3
Improvement in surgical techniques and perioperative care has dramatically changed the natural history of CHD, allowing the survival of up to 95%4 of the patients, resulting in an ever-increasing population of adults reaching fertile age and living with CHD and the consequences of the anomalies and treatments,5 alterations that once would have lead to death at very young ages.
The management of the surviving patients represents a new challenge: 13.6% of the patients with repaired or palliated CHD have associated extracardiac structural malformations,1 in addition to increased risk of arrhythmias, myocardial dysfunction, and neurodevelopmental disabilities, which is potentially the comorbidity with the largest impact on life quality in patients with CHD: they affect 10–50% of the patients, accordingly to the CHD severity.1
The complexity and heterogeneity of CHD have traditionally been attributed to multifactorial etiologies, arising from interactions between multiple genes and environmental factors.2,4 Indeed, it is not easy to precisely define the genetic contribution underlying heart defects, due to the intricacy of the genetic network that controls the organogenesis of the heart. However, many studies point to a major genetic contribution to CHD, such as greater concordance in monozygotic twins, risk of recurrence of related forms of CHD in siblings, and the presence of rare Mendelian forms of heart defects.1
Basis of heart developmentThe heart is the first functional organ to be developed in vertebrate embryos and this process is strictly controlled by a gene regulatory network,2,6 including transcription factors, signaling pathways, microRNAs, and epigenetic factors.
In mammals, three cell lines cooperate in the course of cardiac morphogenesis: cardiogenic mesoderm cells (CMCs), the proepicardium (PE), and neural crest cardiogenic cells (NCCCs).7 The first heart field (FHF) and second heart field (SHF) – which form the major proportion of the ventricular, atrial, and OFT myocardium, the endocardium, the conduction system, and the pulmonary and aortic cushions – are harbored in the cardiogenic mesoderm.7–9 Initially, the FHF forms the heart crescent, which evolves to the heart tube, which is the major contributor to the early left ventricle.
As the heart tube forms, the SHF migrates into the midline and positions itself dorsally to the heart tube, comprising the dorsal-medial aspect of the heart plate, while the FHF comprises the ventral aspect. The FHF differentiates as the cardiac crescent, while the differentiation of SHF is delayed by the inhibitory Wnt signaling that emanate from the midline.2,8 Then, it grows and populates a great part of the OFT, the primary right ventricle, and the atria. Factors secreted from the anterior portion of the heart tube function as chemoattractants to the cells of the SHF, although these mechanisms remain unknown.2,8
Both lineages appear to be controlled by intricate positive and negative signals from pathways such as bone morphogenetic proteins (BMPs), fibroblast growth factors (FGF), Sonic Hedgehog (SHH), Wnt, and Notch signaling pathways.2,7 The early cardiogenic commitment depends on the Nkx2.5 transcription factor expression in mesenchymal cells, as a consequence of the expression of BMP2/4 associated with inhibitors of the Wnt pathway.9,10
Progenitors arising from the PE comprise the epicardium and differentiate as fibroblasts, smooth-muscle vessels, and endothelial cells of the coronaries and some myocytes form the atrioventricular (AV) septum. The interaction between the epicardium and the myocardium is crucial to chamber maturation and ventricular muscle growth.7 This interaction is provided by an extracellular matrix called cardiac jelly, which favors the reciprocal signaling between the outer myocardium and the inner endocardium.2
Finally, the NCCCs originate from the dorsal neural tube and migrate to the third, fourth, and sixth pharyngeal arches, comprising distal OFT and aorticopulmonary ridge smooth muscle cells as well as the autonomic innervation of the heart.7–9 The NCCCs are essential to the maturation and septation of the arterial pole of the heart and contribute to septal and valve formation.6
The heart is the first organ to break the embryonic symmetry as the tube starts its rightward looping, reflecting a global establishment of LR asymmetry, involving the intricate cross talk amongst pathways such as Notch, Nodal, SHH, FGF, and BMP, and finally restricting the Nodal signaling to the left side of the embryo,2,6 through the activity of ciliary cells which generate a sense directional flow of extraembryonic fluid.1,4
The establishment of LR asymmetry is followed by the formation of the endocardial cushions within the OFT and the AV canal that contribute to divide the heart into the four cardiac chambers, starting the division of the OFT into the aorta and the pulmonary tract, and proceeding to valve formation at each end of the heart tube.2,4
Genetic alterations underlying CHDThe majority of CHDs occur as isolated malformations, while 25–30% of them are associated with extracardiac anomalies, and some specific defects are frequently found in association with known genetic syndromes11 as well as several genetic data points contributing to the majority of CHDs,1 although classical Mendelian inheritance patterns are not usually observed.12,13
Major chromosome anomalies have been associated with CHD for more than half a century. Aneuploidies are the earliest identified genetic causes of CHD and the contribution of cytogenetic abnormalities ranges from 9% to 18%.1 The large number of affected genes results in pleiotropic and severe phenotypes, and 98% of the affected fetuses have at least one extracardiac abnormality.1
Newer genetic investigation tools, such as array-CGH, were crucial in revealing the presence of submicroscopic structural anomalies associated with identifiable genetic syndromes, including CHD phenotypes.12,14 Somatic mutations are not a common cause of CHD, but there is a possibility that they may play a role in the disease development in a polygenic or multifactorial setting.14
Chromosome abnormalities and CHDClassic chromosome anomalies detectable by normal standard karyotype include trisomy 21 (Down syndrome, Online Mendelian Inheritance in Man [OMIM] 190685), trisomy 13 (Patau syndrome), trisomy 18 (Edwards syndrome), and monosomy X (Turner syndrome).11 CHD is observed in up to 50% of liveborns with trisomy 21, 60–80% of liveborns with trisomy 13, and 33% of those with monosomy X.1 Each chromosomal abnormality is preferentially associated with specific types of CHD, such as occurring with AV defects and Down syndrome or left ventricle obstructive lesions and Turner syndrome.1,11
Submicroscopic chromosomal anomalies are detectable by fluorescent in situ hybridization (FISH), multiplex ligation-dependent amplification (MLPA), and chromosomal microarrays (CMA). Those techniques increased the knowledge about copy number variations (CNVs): common genomic variations in the population that include deletions and duplications with different genomic consequences. CNVs usually arise from genomic rearrangements at common chromosomal breakpoints due to genomic architecture; they are not necessarily pathological. However, rare CNVs can lead to increase, rupture, or reduction of the expression of one or more genomic elements, leading to pathologies and developmental problems; they are constantly being linked to syndromic and non-syndromic CHD.15,16
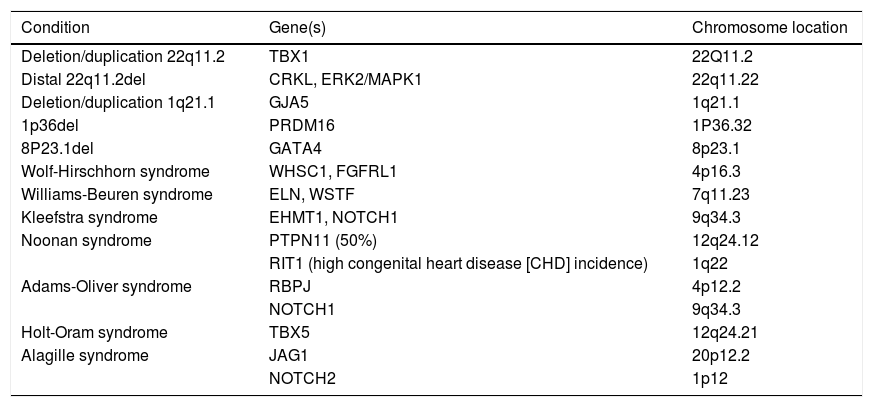
In order to understand the contribution of structural chromosome abnormalities to etiology of CHD, the main genomic disorders (CNV-associated syndromes) related to CHD are detailed below, summarized in Table 1.
Copy number variations (CNVs) associated with congenital heart disease.
| Condition | Gene(s) | Chromosome location |
|---|---|---|
| Deletion/duplication 22q11.2 | TBX1 | 22Q11.2 |
| Distal 22q11.2del | CRKL, ERK2/MAPK1 | 22q11.22 |
| Deletion/duplication 1q21.1 | GJA5 | 1q21.1 |
| 1p36del | PRDM16 | 1P36.32 |
| 8P23.1del | GATA4 | 8p23.1 |
| Wolf-Hirschhorn syndrome | WHSC1, FGFRL1 | 4p16.3 |
| Williams-Beuren syndrome | ELN, WSTF | 7q11.23 |
| Kleefstra syndrome | EHMT1, NOTCH1 | 9q34.3 |
| Noonan syndrome | PTPN11 (50%) | 12q24.12 |
| RIT1 (high congenital heart disease [CHD] incidence) | 1q22 | |
| Adams-Oliver syndrome | RBPJ | 4p12.2 |
| NOTCH1 | 9q34.3 | |
| Holt-Oram syndrome | TBX5 | 12q24.21 |
| Alagille syndrome | JAG1 | 20p12.2 |
| NOTCH2 | 1p12 |
The most common human microdeletion ranging from 0.7 to 3Mbp that affects an estimated 1 in 4000 people, resulting in a broad spectrum of phenotypes characteristic of DiGeorge syndrome (OMIM 188400), velocardiofacial (OMIM 192430) and Shprintzen syndrome (OMIM 182212), encompassing CHD, especially conotruncal abnormalities, palate abnormalities, hypocalcemia, immunodeficiency, characteristic facial features, and neurodevelopmental abnormalities.1
22q11.2 deletion syndrome (22q11del) includes alterations in the T-box transcription factor TBX1, which highlights the significance of the SHF transcription regulation, since TBX1 is a central piece in the proper development of the OFT myocardium4,17 and is expressed solely in the SHF, not in the FHF nor the NCCCs. TBX1 also regulates expression of growth factors, such as fibroblast growth factor 8 (FGF8), which activates the differentiation of NCCCs. Therefore, OFT defects, typically truncus arteriosus, tetralogy of Fallot, and aortic arch anomalies are highly associated with 22q11del, as well as craniofacial defects, including cleft palate,18 and thymic and parathyroid hypoplasia.
Most patients with 22q11del have de novo deletions resulting from non-homologous recombination between low-copy repeats that flank the critical region of the chromosome.
More proximal deletions maintain a phenotype close to DiGeorge syndrome, including immunosuppression, while more distal deletions present incomplete clinical features: genes functionally interacting with TBX1 but located distal to it, such as CRKL and ERK2/MAPK1, have been proposed as the etiology of CHD in more distal deletions, in particular the haploinsufficiency of the MAPK1 gene.11
22q11.2 duplication syndrome (OMIM 608363)Phenotypically similar to the correspondent microdeletion, it is difficult to establish any clear genotype–phenotype correlation for the 22q11.2 microduplication; however, the defects appear to belong to different pathogenic pathways: septal defects and obstructive left ventricle lesions, usually associated with neurological disturbances and growth retardation. The prevalence of CHD in the duplication 22q11.2 is lower compared to the deletion of the same region, but the molecular basis also considers the TBX1 gene as a candidate, as it is overexpressed and may interact with other genes inside and outside the affected chromosomal region.11 The pathogenicity of the microduplication is still hard to define, as the majority of parental carriers have a normal phenotype. However, the notorious prevalence of this microduplication in patients with neurodevelopmental disabilities and the higher occurrence of a second CNV in affected carriers suggest that distal 22q11 microduplications can act as a susceptibility locus for neurodevelopmental disability.19
1q21.1 deletion and duplicationCHD is a major feature of 1q21.1 deletion syndrome (OMIM 612474), with a heterogenous phenotype including mild-to-moderate intellectual disability, microcephaly, and CHD, such as left-side obstructions (40%), septal defects (27%), and conotruncal anomalies (20%).11 There were no notable phenotypic differences among carriers of deletions with different breaking points.20 1q21.1 duplication syndrome (OMIM 612475) is far less common and includes GJA5, described as a susceptibility gene for CHD, notably, tetralogy of Fallot, and has been described as mutated in patients with non-syndromic CHD.4,18,21 The GJA5 gene, which encodes the Cx40 connexin (Cx40),22 a cardiac gap junction cell membrane channel protein that interconnects the cytoplasm of neighboring cells and that is responsible for cell-to-cell conduction of the action potential. The Cx40 connexin is richly expressed in the atrial myocardium and in the atrioventricular conduction system.4 Imbalance in the expression of this connexin is associated with increased propensity for arrhythmias. In addition, patients with mutated forms also show developmental delay and dysmorphic features.21,22
1p36 deletion syndrome1p36Del syndrome (OMIM 607872) is the second most common microdeletion disorder; it is characterized by intellectual disability, epilepsy, dysmorphic features, and metabolic and neuromuscular disorders. Terminal and interstitial deletions are observed with highly variable breakpoints. CHD is present in 50% of the cases, especially in cardiomyopathy, and there is a high prevalence of left ventricle non-compaction.23 The gene that encodes the transcription factor PRDM16 is located inside the critical region of 1p36 syndrome and is linked to non-syndromic left ventricle non-compaction.23,24
8p23.1 deletionDeletions involving the chromosome 8p23.1 range from large deletions including the 8p telomere and detectable by routine karyotyping to small interstitial deletions resulting in different phenotypes, particularly diaphragmatic hernia and CHD.4,24 Cardiac defects are observed in 94% of cases, ranging from isolated septal defects to more complex CHDs, such as tetralogy of Fallot and hypoplastic left heart syndrome.4
The high incidence of CHD is due mainly to the absence or imbalanced expression of the transcription factor GATA4, which is known to play a key role in heart development in humans; the haploinsufficiency of the GATA4 gene has been described as an etiology of non-syndromic CHD in animal models and in families, especially in septal defects.4,11,15 Patients reported with 8p23.1 deletions may have more severe and complex CHD when compared with patients with mutations in the GATA4 gene alone, suggesting that other genes located in the region may play a role in the CHD phenotype.25 Among these genes, haploinsufficiency of the transcription factor gene SOX7 is one of the most likely to exacerbate the effects of GATA4 deletion.25
Wolf-Hirschhorn syndrome (4pter deletion)Wolf-Hirschhorn syndrome (WHS, OMIM 194190) is caused by the loss of the distal portion of the region 4p, with the breaking point usually between 4p15 and 4p16.26 The estimated frequency is around 1:50,000 liveborns.26 The phenotype includes characteristic facial features (known as ‘Greek helmet’ facies, with distinct nose, ocular hypertelorism, short philtrum, high forehead, and arched eyebrows), neurological and growth delay, and seizures. CHD is described in 50% of cases, particularly mild septal defects and arterial ductus persistency, although more severe heart defects have been reported.26,27 The most probable gene implicated in the CHD phenotype is the histone lysine methyltransferase WHSC1, which interacts with the cardiac transcription factor Nkx2.5, especially during cardiac septal formation.4 Another candidate is the FGFRL1 gene, which encodes a member of the fibroblast growth receptor family expressed in the brain, cranial placodes, pharyngeal, arches and heart.
Williams-Beuren syndromeWilliams-Beuren syndrome (WBS, OMIM 194050) is caused by a typical 1.5–1.8Mbp deletion in the 7q11.23 region, involving about 28 genes, affecting 1:7500 to 1:10,000 people. Most patients are heterozygous for a 1.5–1.8Mbp deletion.4 Cardiovascular anomalies are present in 75% of the individuals, usually supravalvar aortic stenosis and pulmonary stenosis, which can be explained by haploinsufficiency of the elastin gene (ELN), causing deficiency or abnormal deposition of elastin in the arterial wall, leading to proliferation of arterial smooth muscle cells and subsequent intimal hyperplasia.28 Point mutations in the ELN gene have been reported in patients with non-syndromic supravalvar aortic stenosis.4
Other CHDs, such as septal defects and tetralogy of Fallot, are described in 6–10% of the patients and cannot be explained by the deletion of ELN gene. Animal models indicates that the deletion of another gene in the 7q11.23 region – BAZ1B, also known as Williams syndrome transcription factor (WSTF) – may account for these defects.4,29WSTF codifies a subunit in three ATP-dependent chromatin remodeling complexes that is crucial for the normal gene transcriptional cascades in the developing heart.29
Kleefstra syndromeKleefstra syndrome (KLEFS1, OMIM 610253) is caused by the microdeletion of the 9q34.3 region or, less commonly, by point mutations in the euchromatic histone lysine methyltransferase 1 (EHMT1) gene. Kleefstra syndrome is a clinically recognizable disease with typical face features (flat face with ocular hypertelorism, synophrys, everted lower lip, macroglossia, and anteverted nares) and CHD in approximately 40% of the patients,4,30 including septal defects, aorta coarctation, pulmonary stenosis, or tetralogy of Fallot.16 Important developmental delay, genitourinary alterations, chronic constipation, and epilepsy are also described.30
Single gene mutation and CHDNext-generation sequencing has opened the door for understanding the genetics of complex diseases such as CHD beyond large structural variation, allowing the identification of mutations that were otherwise unddefinable.1
The controlling network of heart development is vast and intricate, and genetic mutations including gain-of-function and loss-of-function that affects this complex process play a significant role in the genetics of CHD.2
Genes mutated in CHD are usually grouped according to function and involvement in specific pathways, as it clarifies the understanding of these genes in heart formation.
Below, some of the important pathways/mechanisms and associated syndromes related to CHD are mentioned.
Noonan syndrome and RASopathiesRASopathies are a group of syndromes caused by mutations in genes of the Ras-MAPK pathway, which is essential for the cell cycle, with regulatory roles in proliferation, differentiation, growth, and metabolism. Therefore, its dysregulation in this cascade accounts for profound developmental consequences.11 They include Noonan syndrome (NS) and other Noonan syndrome related disorders (NSRD), including cardio-facio-cutaneous syndrome (CFC; OMIM 115150), Costello syndrome (CS; OMIM 218040), and NS with multiple lentigines (NSML; also known as LEOPARD syndrome; OMIM 151100). They are clinically overlapping developmental disorders that share many characteristic features as facial dysmorphism, short stature, and cardiac abnormalities, with phenotypes that are broad and heterogeneous, and differential diagnosis among them can be difficult.31
Noonan syndrome is one of the most common genetic syndromes associated with CHD, with an estimated prevalence of 1:1,000 to 1:2,500 liveborns. It is a clinically heterogeneous disorder inherited by autosomal dominant trait.13 The clinical features include short stature, dysmorphic features (such as triangular face, hypertelorism, low-set ears, and ptosis), and lymphatic, hematologic, skeletal, and ectodermic defects. In addition, patients may present variable neurologic impairment, ranging from moderate intellectual disability to superior skills; however, children with severe heart disease tend to present lower cognitive abilities.30 Hearing loss, incoordination, mood disturbances, hyperactivity, and attention deficit have also been described.11,13,32
CHD occurs in 60–90% of patients with RASopathies, with less occurrence in NSML. The most frequent cardiac abnormalities are pulmonary valve stenosis, atrioventricular septal defect (AVSD), septal defects, and hypertrophic cardiomyopathy.8,11,31
Different genes have been identified as responsible for the phenotype of Noonan syndrome or correlated conditions. Missense mutations in the PTPN11 gene – located at 12q24.1 region – are responsible for approximately 50% of the cases. Another twelve genes are implicated and together with PTPN11 account for approximately 90% of affected cases: KRAS, SOS1, RAF1, NRAS, BRAF, SHOC2, PPP1CB, CBL, RRAS, RIT1, LZTR1, and SOS2. Most of these genes encode for proteins that implicated in the RAS-mitogen-activated protein kinase (MAPK) signaling pathway. The RAS/MAPK pathway is an important signaling cascade that allows cells to properly respond to multiple extracellular stimuli, including growth factors, hormones, and cytokines, controlling virtually all the cellular processes. The majority of these mutations lead to an increase of signal transduction down this pathway, causing continuous MAPK activation during development.13,33SOS1, RIT1, and RAF1 are the genes most often mutated, and the prevalence of CHD in patients with mutations in the RIT1 gene is particularly high (90%).11,13
AVSDs are especially found in patients with mutations in the PTPN11 and RAF1 genes, and partial AVSD associated with left-sided obstructions, pulmonary valvar stenosis, or hypertrophic cardiomyopathy should be regarded as markers for Noonan or correlated syndromes.11
Adams-Oliver syndromeAdams-Oliver syndrome (AOS1, OMIM 100300) is a rare developmental disorder characterized by aplasia cutis congenita of the scalp vertex a terminal transverse limb defects, with high intra- and inter-familial variability.11,34 Cardiovascular malformations occur in 13–20% of the patients, and different anatomic types have been reported: left-sided obstructive lesions, septal and conotruncal defects, and tricuspid atresia. Left-sided lesions are prevalent, occurring at multiple levels.34
The classic genes implicated in Adams-Oliver syndrome include ARHGAP31, DOCK6, RBPJ, and EOGT, and only 9% of the patients with these mutations have CHD; in particular, septal defects.34
The RBPJ gene was proposed as a candidate for Adams-Oliver syndrome with more complex CHD, since it codes for a highly preserved protein that coordinates the transcriptional activation of the Notch pathway target-genes, being an important key to the mesenchymal cell formation, skeletal, vascular, epidermis, and hair-follicle formation.35 Variants of the NOTCH1 gene, belonging to the Notch signaling pathway have been shown to be related to Adams-Oliver Syndrome with CHD, and it has been proposed that the limb and scalp defects are secondary to the vasculopathy caused by NOTCH1 haploinsufficiency.11 The NOTCH1 gene has not only been implicated in non-syndromic CHD but is also known to be essential for the transformation of epidermal cells to migratory mesenchymal cells and definition of the valvar territory, and it is broadly expressed in the OFT.35
Holt-Oram syndromeHolt-Oram syndrome (HOS, OMIM 142900) affects 1:100,000 people and can be sporadic or inherited as an autosomal dominant disease caused by either nonsense or frameshift mutations in the TBX5 gene,13,16 or even duplications involving this gene on the 12q region.35 TBX5 is a transcription factor that is a known key regulator of heart organogenesis, especially when in combination with other transcription factors, such as Nkx2.5 and GATA4.2,13,17
This syndrome is characterized by upper limb malformations and CHD, marked septal defects, and conduction diseases. Upper limbs abnormalities are always present, involving structures derived from the radial ray, and are most commonly bilateral and asymmetric, ranging from subclinical radiological findings to phocomelia. Heart defects are typically septal, but more complex heart diseases have been described. Cardiac conduction abnormalities are also commonly found. No correlation can be made between the severity of limb abnormalities and heart malformation.36
Alagille syndromeAlagille syndrome (ALGS1, OMIM 118450) is a multisystem disorder with estimated prevalence of 1:70,000 newborns, which involves the heart, liver, eyes, face, and skeleton. CHD is present in 90% of the cases; involvement of the pulmonary OFT is the most common type of CHD described, among which pulmonary valvar and/or arterial stenosis and tetralogy of Fallot are usually mentioned. The paucity of the interlobular bile ducts and consequent cholestasis is also an important clinical feature.13
The vast majority of the Alagille syndrome patients (>90%) have mutations in the JAG1 gene, which codifies a Notch-signaling ligand. Selected cases (<1%) have mutations in the NOTCH2 gene.37 The JAG1 gene is strongly correlated with cardiovascular malformations and underlies non-syndromic CHD, most notably tetralogy of Fallot.1
As CHD may be an isolated manifestation of Alagille syndrome, patients with family history of Tetralogy of fallot or those with branch pulmonary artery stenosis or hypoplasia should be entitled to genetic testing, even if other phenotypic features are absent.1,13
Genes involved in epigenetic controlNew approaches are been used in the pursuit of understanding both the etiology and phenotypic variability of complex genetic diseases. The study of epigenetics – genomic changes that do not involve DNA sequence modifications – suggests that chromatin structure and/or epigenetic disturbances may lead to changes in the transcription of multiple genes and metabolic pathways that may play a key role in CHDs.
Several studies demonstrate the importance of various mechanisms of epigenetic regulation during cardiogenesis.38 Alterations of DNA methylation, especially CpG islands close to transcription factors, have been identified in patients with cardiac malformations.38 Studies with next-generation sequencing have identified significant enrichment for mutations in genes involving histone modification in patients with CHD, especially H3K4, H2K7, H3K9, and H3K27, suggesting that histone modification may be significant in the pathology of isolated disease.1,4
Investigation of chromatin remodeling in model organisms has shown that dynamic modification of chromatin structure plays an important role in the regulation of gene expression during heart development.13De novo mutations affecting chromatin regulation genes contribute to about 3% of the CHDs. In addition, chromatin regulation genes encompass about 600 genes that orchestrate the dynamic gene expression by altering epigenetics factors or catalyzing alterations in the chromatin structure.1 Therefore, genes that encodes proteins which modify or bind to histones have been implicated in the etiology of syndromes that cause CHD, such as the following:
Kabuki syndromeKabuki syndrome (KABUK1, OMIM 147920) is genetic disorder that affects 1:30,000 newborns and causes developmental delay, facial dysmorphisms, and left-sided obstructive lesions, which initially raised a suspicion of involvement of the X chromosome, although septal and conotruncal defects can also be detected.11 In fact, notwithstanding the identification of the MLL2 gene – a histone methyltransferase – as the primary cause of the Kabuki syndrome through whole genome sequencing, de novo partial or complete deletion of X chromosome genes, which encode the histone modifiers KMT2D and KDM6A that interacts with MLL2 gene may also result in a Kabuki syndrome phenotype.11,13
CHARGE syndromeCHARGE (OMIM 214800) is an acronym that stands for iris Coloboma, Heart malformation, choanal Atresia, Retarded growth and development, Genital hypoplasia, and Ear anomalies and deafness, although other malformations and behavioral alterations may be present and the diagnostic criteria have been refined several times.39 It affects 1:8000 to 1:10,000 newborns and around 70% of the patients have CHD; about half of them have major conotruncal defects, such as tetralogy of Fallot and double outlet of the right ventricle, albeit other OFT like hypoplastic left heart syndrome are also described.13,39 More than two-thirds of the cases are caused by nonsense or frameshift mutation of the CHD7 gene, which encodes a chromatin modifier protein, although alterations in the semaphorin gene (SMA3E) may result in similar phenotype.40 The CHD7 protein is essential for neural crest migration, which can explain the high frequency of OFT defects.13,39
Koolen-De Vries syndromeKoolen-De Vries Syndrome (KDVS, OMIM 610443) is caused by deletion of the 17q21.31 locus or mutation of the KANSL1 gene, located in the aforementioned locus, and is characterized by severe intellectual deficit, hypotonia, seizures, and facial dysmorphisms. CHD is present in 27% of cases, especially septal defects, although pulmonary stenosis may also be described.1,41 Recent studies have identified that KANSL1 plays a role as a modifier gene in 22q11.2DS patients.42
Non-syndromic CHDThe vast majority of CHDs – about 70% – occur as isolated malformations,11,12,43 including the most complex: tricuspid atresia, great artery transposition, hypoplastic left heart syndrome, and pulmonary atresia. Several new genes with Mendelian inheritance have been identified, and studies of families affected have not only shed a light on the inheritance patterns, but also have been essential to the understanding of complex heart organogenesis, since the genes etiologically linked to CHD directly impact the embryologic development and may also play a role in heart regulation throughout life.12 Next-generation sequencing (NGS) technology has opened the door for discerning the importance of de novo variants without clear Mendelian inheritance, variants of reduced penetrance, and somatic alterations, among others.12,13
Most of identified mutations are family-specific and cannot account for the common causes of CHD, but it is possible that multiple variants may play a role in the disease development in a polygenic setting, although the interpretation of these variants can be very challenging and it is not always possible to establish their pathogenicity: these associations can be highly significant from a statistical and research perspective, but with low clinical relevance.14
In many families and individuals with CHD, variations in genes expressed during heart formation are present with different profiles of inheritance, suggesting a continuum between Mendelian and complex forms of diseases, apart from single-gene disorders as exemplified below and listed in Table 2.14
Genes associated with non-syndromic congenital heart diseases.
| Condition | Gene | Chromosome location |
|---|---|---|
| Septal defects; abnormal conduction; tetralogy of Fallot; left heart hypoplasia | NKX2-5 | 5q35.1 |
| Tetralogy of Fallot; double outlet of right ventricle; ventricular septal defects | NKX2-6 | 8p21.2 |
| Septal defects; double outlet of the right ventricle | TBX20 | 7p14.2 |
| Septal defects; double outlet of the right ventricle; tetralogy of Fallot | GATA4 | 8p23.1 |
| Tetralogy of Fallot; familial atrial fibrillation | GATA5 | 20q13.33 |
| Outflow tract defects | GATA6 | 18q11.2 |
| Heterotaxy | ZIC3 | Xq26.3 |
| Atrial isomerism, D-transposition of great arteries | PITX2 | 4q25 |
| Familial atrial septal defect | MYH6 | 14q11.2 |
| Tetralogy of Fallot | JAG1 | 20p12.2 |
| Left heart hypoplasia | NOTCH2 | 1p12 |
The NK2 family are homebox-containing genes that play crucial roles in heart development, regulating essential processes such as the spatial and temporal gene expression.8,9,44 The NKX2-5 gene is expressed both in the first and second heart fields as one of the earliest markers of cardiomyogenic differentiation and is central to the cardiac regulatory hierarchy.2 Several mutations have been described, leading mostly to septal defects and atrioventricular conduction abnormalities,2 but more complex CHDs, such as tetralogy of Fallot and left heart hypoplasia, have also been described.45 Recent studies have focused on the regulatory region of the NKX2-5 gene, proposing that these non-coding variants may enhance transcription and alter the network that controls cardiac morphogenesis. It has also been postulated that these mutated versions can bind to promoters of non-specific genes and allow co-factors to induce a stronger effect than usual, which can explain the wide variations of phenotypes in affected individuals.45
NKX2-6 partially overlaps with NKX2-5 in temporal and spatial expression profiles and functional characteristics during embryogenesis. Loss of function mutations of NKX2-6 have been identified in patients with tetralogy of Fallot, double outlet of the right ventricle, and ventricular septum defects.2
Mutations in the TBX familyThe toolbox (TBX) transcription factor family is a group of six proteins sharing a highly conserved DNA-binding domain and with significant role in the development of cardiac progenitor cells – especially in the second heart field – as well as in the patterning of the chambers and OFT.2,17
The TBX1 gene is expressed in the pharyngeal mesenchyme and epithelium and is a major genetic determinant of cardiac and craniofacial disorders, being included in the set of genes deleted in 22q11del syndrome. Mutations in the TBX5 gene have been associated with Holt-Oram syndrome, as described. There are very few cases of isolated CHD related to mutations in those two genes.44
However, mutations in the TBX20 gene have been associated with atrial and ventricular septal defects and aberrant valvulogenesis: TBX20 is required in endothelial lineages for septation, regulating versican, an extracellular matrix proteoglycan, and the proliferation and differentiation of cardiomyocytes in the septa.4,46 Mutations in TBX20 increase the susceptibility to double outlet of the right ventricle in humans, and also have been causatively linked to dilated cardiomyopathy.2
Mutations in the GATA familyThe family of the GATA zinc-finger transcription factors comprises six members: GATA1 to GATA6, which bind to the base sequence (A/T)GATA(A/G) in the regulatory region of numerous genes. Most tissues of mesodermal or endodermal origin express at least one of the following: GATA4, GATA5, or GATA6, and all three of them are present in the precardiac mesoderm.2,7 Experiments in animal models have shown that silencing the GATA genes can result in CHDs ranging from valvoseptal defects to acardia.44
GATA4 is the most investigated member, and also one of the earliest transcription factors expressed in developing heart cells.17,47 Decrease in the expression of the GATA4 gene leads to various forms to CHD, such as atrioventricular septal defect, double outlet of right ventricle, and familial forms of tetralogy of Fallot.2,17,47
GATA5 can promote cardiomyocyte fates from murine embryonic stem cells.6 Little is known about GATA5 mutations in humans, but three heterozygous mutations have been identified in families with anatomical heart malformations or familial atrial fibrillation2,47 and in sporadic tetralogy of Fallot.2
GATA6 is highly expressed not only in the developing heart – precardiac mesoderm, heart tube – but also in adult cardiomyocytes in human ventricles, and atrial and vascular smooth cells.2,47 Deletion of GATA6 in neural crest-derived smooth muscle cells may result in OFT defects, such as interrupted aortic arch and persistent truncus arteriosus, phenotypes associated with severely decreased expression of SEMA3C.17,47 Endocardial cushion formation is also impacted by GATA6, thus mutations in this gene have been implicated in non-syndromic tetralogy of Fallot and atrioventricular septal defect.2
ZIC3 mutationsThe ZIC3 gene encodes for a zinc-finger transcription factor that is implicated in LR axis development, thus known as heterotaxy gene. Located in the X chromosome, loss of function mutations in Zinc3 lead to X-linked heterotaxy and isolated CHD,2,14 such as D-transposition of the great arteries and double outlet of the right ventricle.48
PITX2 mutationsThe PITX2 gene belongs to the pituitary homebox family of transcription factors, which play a role in both DNA and RNA binding, and consists of three isoforms: PITX2a, PITX2b, and PITX2c. The LR asymmetry of the heart depends on the expression of PITX2 via nodal pathway on the left side, with activation of wnt-dependent cell cycle pathways downstream, and its repression on the right.2,49 PITX2 loss of function of any isoform causes severe atrial isomerism, double inlet left ventricle, D-transposition of great arteries and persistent truncus arteriosus.49
Genes encoding components of the cardiac sarcomereSarcomeric genes are largely recognized as candidates for diverse familial cardiomyopathies, but a few genes have also been linked to structural heart diseases.4,50 Mutations in the MYH6 (myosin heavy chain 6) gene were identified in familial forms of atrial septal defect (ASD), and molecular regulation involves transcription factors such as GATA4 and TBX5. The incidence of ASD may also be overrepresented in noncompaction of left ventricle, caused by MYH7 mutation,4 which is also related to Ebstein's anomaly.50,51
Notch pathway genesThe Notch signaling is a highly conserved pathway mediating local intercellular communication and regulates cell patterning and is crucial in organs with complex architecture.34 Notch pathway is particularly important during atrioventricular canal and OFT formation and morphogenesis, and mutations of the genes involved in humans result in very specific cardiovascular development impairments and syndromes, such as Alagille's or Adams-Oliver.1,34JAG1 mutations can be implicated in isolated CHD, notably Tetralogy of Fallot.1NOTCH1 mutations have been associated, within a single family, with a range of CHD form bivalvular aortic valve to hypoplastic left heart syndrome. GALNT11 have been linked to human heterotaxy.1
Cilia genesThe cilia serve to multiple functions, including signaling, extracellular fluid propulsion and cell cycle control, and mutations in these genes may cause diverse human disorders with pleiotropic phenotypes. In the heart development, the best understood role for cilia is the establishment of the LR asymmetry, thus mutations affecting the ciliary motility can result in heterotaxy and CHD.1 In animal models, mutations in genes coding components of the dynein motor complex (Dnah11/LRD and Dnah5) result in cardiac and visceral LR abnormalities.1 Not surprisingly, 12.1% of patients with primary ciliary dyskinesia present some form of laterality defect, with or without cardiac defects.52
ConclusionThe development of the heart is extremely complex and demands interactions among countless molecular and epigenetic factors. As the care of the patient with CHD evolves and allows them to grow and reproduce, the necessity of understanding the genetic role arises, particularly in sporadic CHD. For bedside management, the recognition of the genetic alterations underlying heart disease may be helpful in defining prognosis and anticipating complications, such as systemic inflammatory response, arrhythmias, and early heart failure.
With NGS technologies, comprehension of CHD biology has expanded rapidly, but there are still many questions to be answered, as the genetic underpinnings of more than 50% of the cases remain unknown. The extreme genetic and clinical heterogeneity and poor genotype-phenotype correlation makes this path even more challenging.
FundingThis work was supported by MCT/CNPq, CAPES, and FAP-DF.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Saliba A, Figueiredo AC, Baroneza JE, Afiune JY, Pic-Taylor A, Oliveira SF, et al. Genetic and genomics in congenital heart disease: a clinical review. J Pediatr (Rio J). 2020;96:279–88.




