

Clinical use of microarray-based techniques for the analysis of many developmental disorders has emerged during the last decade. Thus, chromosomal microarray has been positioned as a first-tier test. This study reports the first experience in a Chilean cohort.
MethodsChilean patients with developmental disabilities and congenital anomalies were studied with a high-density microarray (CytoScan™ HD Array, Affymetrix, Inc., Santa Clara, CA, USA). Patients had previous cytogenetic studies with either a normal result or a poorly characterized anomaly.
ResultsThis study tested 40 patients selected by two or more criteria, including: major congenital anomalies, facial dysmorphism, developmental delay, and intellectual disability. Copy number variants (CNVs) were found in 72.5% of patients, while a pathogenic CNV was found in 25% of patients and a CNV of uncertain clinical significance was found in 2.5% of patients.
ConclusionChromosomal microarray analysis is a useful and powerful tool for diagnosis of developmental diseases, by allowing accurate diagnosis, improving the diagnosis rate, and discovering new etiologies. The higher cost is a limitation for widespread use in this setting.
O uso clínico de técnicas baseadas em microarrays para a análise de transtornos de desenvolvimento tem surgido durante a última década. Assim, o microarray cromossômico tem sido posicionado como um teste de primeiro nível clínico. Relatamos a primeira experiência em uma coorte chilena.
MétodosPacientes chilenos com atraso de desenvolvimento e anomalias congênitas foram estudados com um microarray de alta densidade (CytoScan™ HD Array, Affymetrix, Inc., Santa Clara, CA, EUA). Pacientes tiveram estudos citogenéticos anteriores, ou um resultado normal ou de uma anomalia não bem caracterizada.
ResultadosForam analisados 40 pacientes selecionados por dois ou mais critérios, incluindo: anomalias congênitas maiores, dismorfismo facial, atraso de desenvolvimento e deficiência intelectual. Uma variante do número de cópia (CNV) foi encontrada em 72,5% dos pacientes, enquanto que uma CNV patogênica foi encontrada em 25% dos pacientes e uma CNV de significado clínico incerto foi encontrada em 2,5% dos pacientes.
ConclusõesA análise cromossômica microarray é uma ferramenta útil e poderosa em transtornos de desenvolvimento, permitindo um diagnóstico preciso, melhorando a taxa de diagnóstico, e descobrindo novas etiologias. O custo mais elevado é uma limitação para um uso difundido em nossa realidade.
Major congenital anomalies affect two to three of every 100 live newborns, and are a leading cause of infant mortality and disability.1,2 Although most are isolated and multifactorial in origin, patients with multiple abnormalities require an assessment to identify an underlying genetic cause.
In recent years, the etiological study of developmental disorders has been enriched with the clinical use of microarray-based techniques. In developed countries, molecular karyotyping or chromosomal microarray (CMA) is considered the first-line technique for the analysis of patients with multiple congenital anomalies, nonsyndromic developmental delay/intellectual disability, and autism spectrum disorders.3–7
In contrast, in developing nations such as Latin American countries, detection of chromosomal anomalies is still performed mainly by conventional cytogenetic techniques. GTG (G-bands by trypsin using Giemsa) banding karyotyping in lymphocytes has been mainly used to identify chromosomal abnormalities with a resolution equal or greater than 5-10 megabases (5-10 Mb).8–11 Fluorescent in situ hybridization (FISH) is available for a limited number of diseases caused by chromosomal microdeletions/microduplications and has a resolution of 2-5 Mb in metaphase and between 50-150 Kb in interphase nuclei.8,9,11–13 Other molecular techniques have been developed to look for small microdeletions/microduplications, such as multiplex ligation-dependent probe amplification (MLPA).14 In contrast to these conventional techniques, CMA has a higher resolution, which reaches up to 50 Kb, a ten times higher resolution than conventional karyotyping.13,15 It seeks genetic imbalances (gains or losses of chromosomal segments) across the genome and has allowed the identification of new syndromes that are not readily detected by the methods described above.16–18 The discovery of normal variation as copy number variations (CNVs) poses a challenge for the clinical interpretation.15
Whilst diagnostic studies for individuals with congenital anomalies or intellectual disability based on conventional cytogenetics have a diagnostic yield close to 3%, CMA has a yield of around 15% to 20%, over five timesgreater than G-banded karyotype,6 justifying its use as a first line diagnostic test for patients with an unknown clinical diagnosis. It is estimated that CMA alone is capable of detecting over 99% of all karyotype abnormalities.5
This report presents the authors’ pioneering experience in the use of CMA in a cohort of Chilean patients with multiple congenital anomalies without etiological diagnosis.
MethodsPatientsForty patients were selected from the Genetic Clinics at Hospital Padre Hurtado (Santiago, Chile), between May of 2012 and November of 2012.
This study included patients who had at least two of the following clinical features: major congenital anomalies (MCAs), facial dysmorphism (FD), developmental delay (DD), or intellectual disability (ID). All patients lacked a definite cause for the disorder.
Of all patients, 36 had a normal karyotype, two patients had an uncharacterized small additional marker chromosome (sSMC), one had a derivative chromosome, one had an inherited Robertsonian translocation, and one patient had monosomy X, but with unusual additional features.
The local ethics committee approved this study, and written informed consent was obtained from all patients and/or parents/guardians.
Sample ProcessingGenomic DNA was purified from peripheral blood mononuclear cells with AxyPrep Blood Genomic DNA Miniprep Kit (Axygen Biosciences, Union City, CA, USA) following manufacturer's instructions. Genomic DNA of each patient was hybridized with the CytoScan™ HD Array (Affymetrix, Inc., Santa Clara, CA, USA) according to manufacturer's instructions. This is a custom high-density comparative genomic hybridization array with almost 2.7 million of genetic markers, includes 700,000 single nucleotide polymorphism (SNP) markers and over 1.9 oligonucleotide non-polymorphic probes for CNV detection.
Data AnalysisArray data were analyzed using the Affymetrix® Chromosome Analysis Software Suite (ChAS) v.1.2.2 (Affymetrix, Inc., Santa Clara, CA, USA) based on the reference genome sequence of the UCSC Genome Browser hg19, Feb. 2009 (GRCh37/hg19). The authors analyzed CNVs over 400 Kb as recommended.6 With this higher-resolution platform, it was possible to evaluate smaller CNVs, but no patient had clinically relevant abnormalities between 100 and 400 Kb, so this limit was kept for purposes of this report. CNVs over 400 Kb were categorized by clinical significance as CNV of clear clinical relevance (group 1), CNV of unclear relevance or uncertain significance (group 2), or benign or polymorphic CNV (group 3), using the publicly available databases ISCA (International Standard Cytogenomic Array),19 DGV (Database of Genomic Variants),20,21 OMIM (Online Mendelian Inheritance in Man),22 DECIPHER(Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources)23,24 and ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations).25
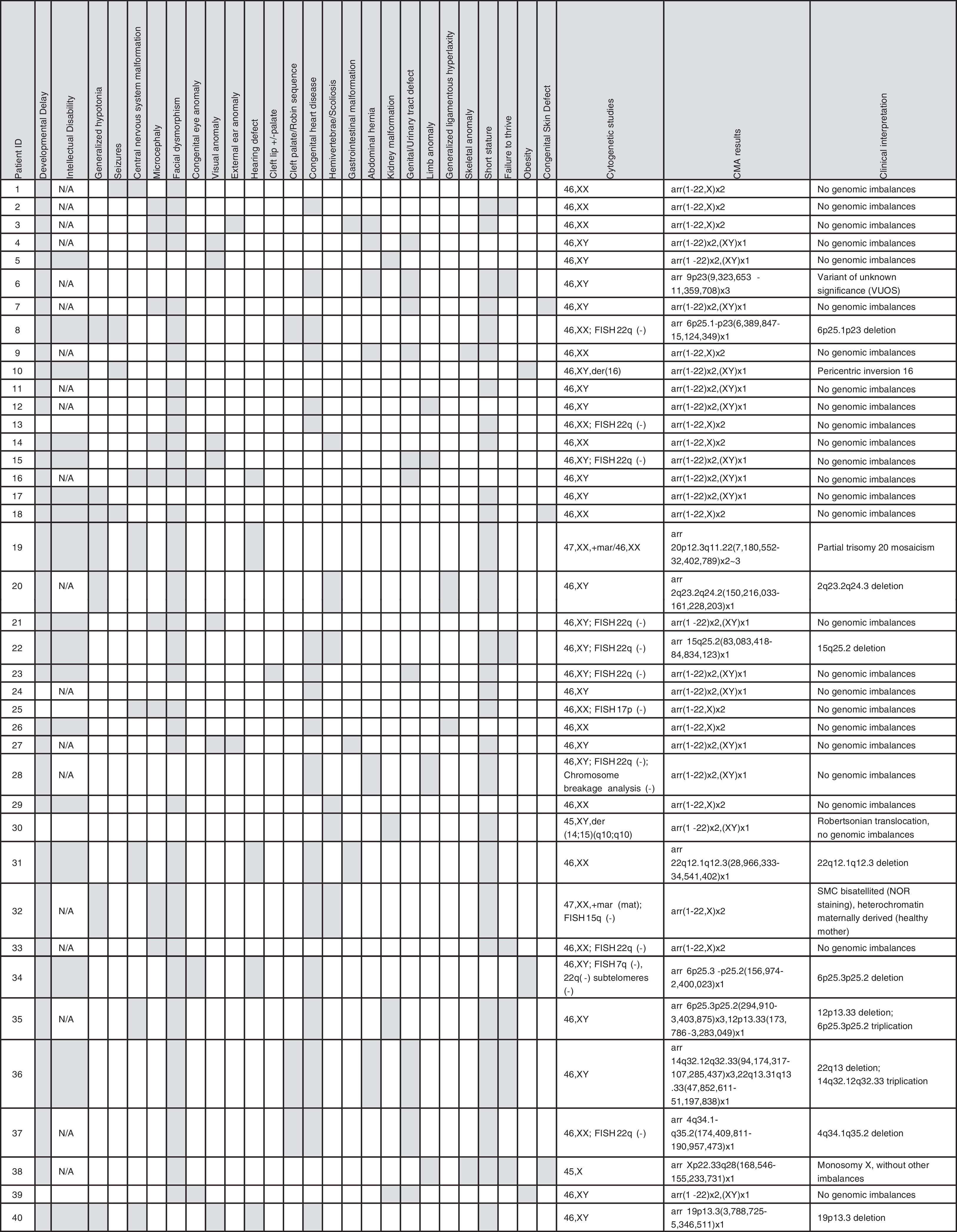
ResultsOf the 40 patients analyzed, 16 (41%) were female. Ages ranged from 1 month to 25 years, with a median age of 4.2 years. As selected, the vast majority of the patients have multiple anomalies, including structural and functional developmental disorders. Clinical details are summarized in Table 1.
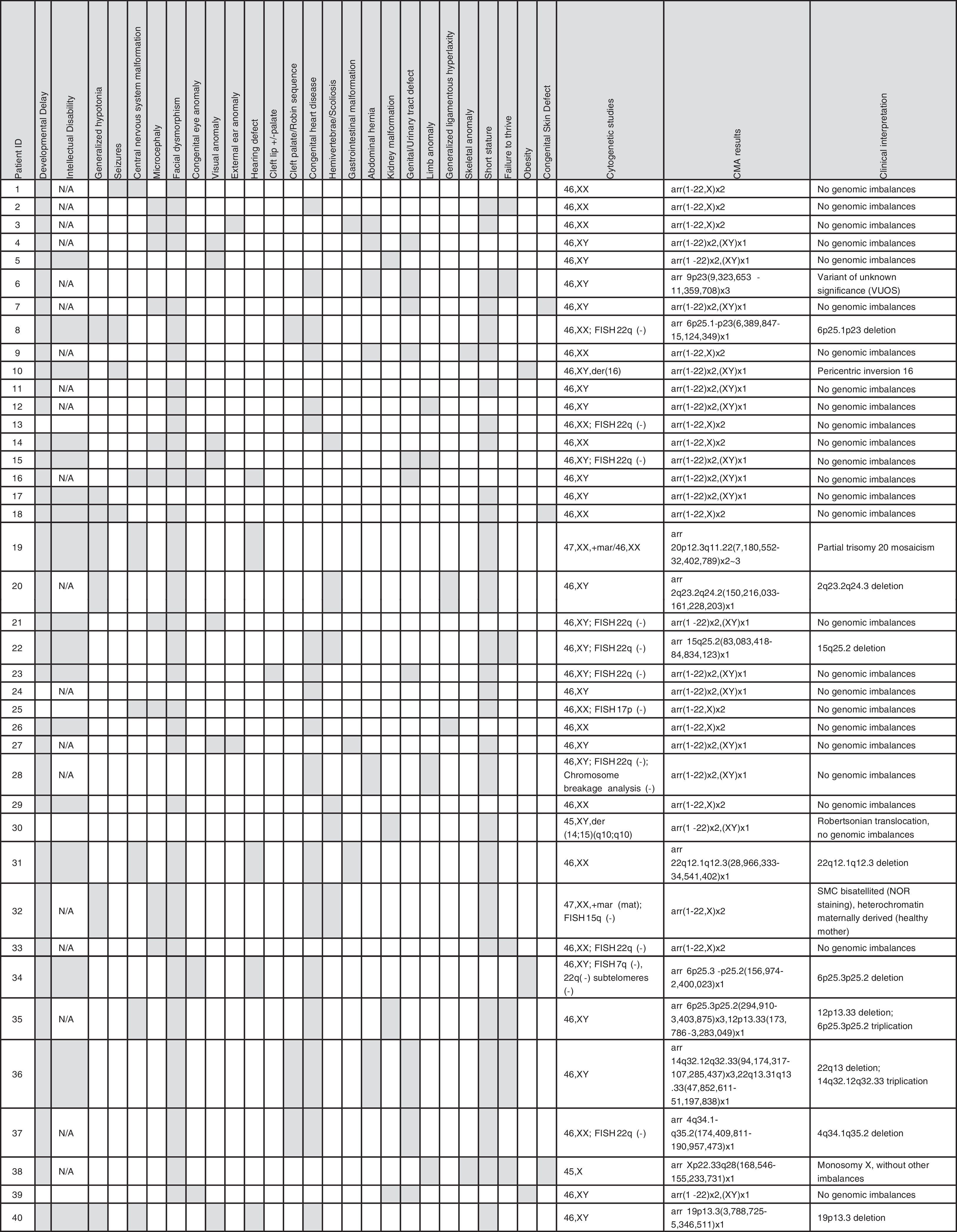
Fifty-five CNVs over 400 Kb were observed in 29 of 40 patients (72.5%), ranging between 0 and 4 CNVs per patient. Of the total, 11 were losses and 44 were gains (Fig. 1A). The size of chromosomal imbalances ranged from 420.9 Kb to 25.2 Mb. The latter corresponded to one patient with an sSMC detected by karyotype.
. B, each patient may have CNV of more than one group at a time. CNVs were categorized by clinical significance as CNV of clear clinical relevance (group 1), CNV of uncertain significance (group 2), or benign or polymorphic CNV (group 3). CNVs, copy number variants; G, gains; L, losses.")
Characterization of CNVs larger than 400 Kb in Chilean cohort.
A, among 40 patients, CNVs were not found in 11 individuals. In the remaining 29 patients, 55 CNVs were found (11 losses and 44 gains). B, each patient may have CNV of more than one group at a time. CNVs were categorized by clinical significance as CNV of clear clinical relevance (group 1), CNV of uncertain significance (group 2), or benign or polymorphic CNV (group 3).
CNVs, copy number variants; G, gains; L, losses.
These 55 CNVs were classified into three groups based on their clinical interpretation.
According to the classification adopted (Fig. 1B), 21.8% belonged to group 1 (clear clinical relevance related to the phenotype), 1.8% to group 2 (unclear relevance), and 76.4% to group 3 (benign or polymorphic). Losses were predominant in group 1, while gains were predominant in group 3.
Diagnostic yieldIn Group 1, two patients had two terminal chromosome imbalances, each probably derived from unbalanced cryptic translocations. Seven patients had a microdeletion and one patient had a mosaic partial trisomy 20 (one of the patients had an sSMC). More details are summarized in Table 1.
In Group 2, only one patient had a variant of uncertain significance (VOUS).26 This patient had a 2 Mb triplication at 9p23 [arr 9p23(9,323,653-11,359,708)x3], but with the information available in the databases and in biomedical literature, a definitive pathogenic effect could not be ruled out or assigned.
The authors failed to find true genetic imbalances in the second patient with an sSMC and the patient with a derivative chromosome 16. In the patient with the Robertsonian translocation, no genomic imbalance that explained the phenotype was found. Finally, in the patient with monosomy X, the deletion of one X was confirmed, but additional genomic imbalances were not found (Table 1).
Clinical interpretationThis study found a pathogenic CNV that was not previously observed by (n = 9) or not well characterized (n = 1) by conventional karyotype in ten of 40 patients of group 1 (25.0%). In addition, a known cytogenetic anomaly in one patient was corroborated (monosomy X).
If only the patients with normal karyotype are considered (35 patients), the diagnosis rate increased to 28.5%.
Additionally, this study found a CNV of uncertain clinical significance or VOUS (group 2) in one additional patient. This patient is waiting for additional testing and follow up to define the clinical relevance of this finding. It is planned to study both parents with FISH and/or MLPA.
DiscussionThis is the first report of the CMA testing in a cohort of Chilean patients with developmental disabilities, considering that there are few studies of the clinical use of chromosomal microarrays in Latin America, as experiences of individual cases are the norm. Within South America, a similar experience was reported with a group of patients from Brazil.27,28 Those authors analyzed 95 syndromic patients with normal karyotypes and reported a diagnostic yield of 17%.28
The present study detected over 25% pathogenic alterations in this cohort, which is in the upper range that has been reported in the literature. In patients with syndromic and nonsyndromic developmental delay/intellectual disability with normal karyotype/FISH, meta-analysis shows a diagnostic yield of 7.8%-13.8%, ranging from 5% to 50%.5,29 This is largely explained by heterogeneity in the design of studies, especially in patient selection, previous testing realized, and array platforms used. In the present case, the high rate of diagnosis can be explained by the fact that the studied cohort was relatively small, had the bias of very selected patients, many of whom have remained for long time without diagnosis, and finally, used a high-density platform.
Over time, different online public databases have collected phenotypic and genomic information of thousands of anonymous patients, which allow elucidating the vast majority of CNVs as benign or polymorphic, without further analysis. In fact, the majority of this study's findings were polymorphic CNVs (copy number polymorphisms, [CNPs]), corroborated in those cytogenomic databases and from the authors’ own cohort data. Recurrent CNVs over 400 Kb were observed in half of these patients, mainly involving the following chromosomal loci: 10q11.22, 14q32.22, 16p11.2, 17q21.31, Yq11.223, and Yq11.23. Only one case that had a VOUS required further analysis to determine possible pathogenicity. This patient was one year and 11 months, with DD, language delay, hearing deficit, inguinal hernia, and short stature. He had a 46,XY karyotype and the array showed a 2 Mb gain in chromosome 9p23, including partial triplication of one gene: PTPRD. The protein Ptprd is a receptor-type protein-tyrosine phosphatase, is expressed in certain regions of the brain, such as the hippocampus, and could have a role in learning and memory30 and the synaptic organization.31 No phenotype has been assigned to the full triplication of this gene.
Although CMA is a very robust and reliable technique, it has limitations: it is unable to detect balanced genomic abnormalities such as inversions, reciprocal, and Robertsonian translocations. Depending on the platform used, low-level mosaicism and some polyploidies cannot be detected. When CMA technique is used as the first line of study, it has been described that this situation can occur in 0.78% of cases.5 Finally, CMA does not give information on position of the rearrangement, FISH is often used as complementary method to identify possible rearrangements with implications for genetic counseling.7,32
In the present cohort, it was found that three patients with previously known cytogenetic abnormalities resulted in a normal molecular karyotype. As expected, the patient who is carrier for a Robertsonian translocation had a normal/balanced aCGH result. In the case of one of the two patients with sSMC, the coverage of the array, the genomic sequences involved, and the size sSMC may explain why that genetic material were undetected by this method.19 Thus, this small bisatellited chromosome likely corresponds to highly repetitive sequences typical of acrocentric chromosomes not included in the array that was demonstrated by nucleolus organizer region (NOR) banding. In the patient with a derivative chromosome 16, with an abnormal banding pattern and morphology (more metacentric than usual), a pericentric inversion is the most plausible explanation.
Thus, karyotype remains more suitable to evaluate potential carriers of chromosomal rearrangements, couples with recurrent miscarriage, or patients with a distinctive aneuploidy phenotype. However, FISH is more suitable if a specific microdeletion syndrome is highly suspected.7
Chromosome microarrays are a highly accurate, robust, and high-throughput method. This study used a combined oligonucleotide-based array plus SNP array, which has many advantages, where the SNP array significantly improves the accuracy and sensitivity of the CNV detection and mosaicism, also allowing the detection of copy-neutral variants.33 In the case of the chip used in this report (Cytoscan HD, Affymetrix), the platform chemistry and its algorithms analyze the oligonucleotide and SNP probes independently, and thus each CNV can be detected and confirmed at the same time, without requiring any further confirmation.32,33 In this case, instead of a confirmation of the array finding, FISH analysis allows for determination of the type of rearrangement.32
A limitation for widespread use of molecular karyotype is the high cost of the test. In Chile, CMA is approximately four to seven times more expensive than a karyotype and/or FISH, and is currently not covered by health insurance. However, to reach an early diagnosis in selected patients with multiple congenital anomalies and/or global developmental delay, it can avoid unnecessary testing (the “diagnostic odyssey”) and allow focusing on specific issues, which in the long term can be cost-effective.34–38 It may be expected that costs will decrease over time, enabling its more widespread use. Finally, it should be mentioned that there are other platforms that have lower resolution but at a relatively reduced cost.
Recognizing its limitations, there is growing evidence of the clinical impact of this technology. In many patients, a definitive diagnosis can impact not only on information and counseling for their family environment,39 but also on active surveillance in search of possible complications, among other types of medical interventions.37,38 The present data show the usefulness of CMA, allowing improved diagnostic capability with remarkable precision and optimization of the management and supervision of health in this group of patients with special needs.
FundingGrant support from Child Health Foundation in Birmingham, Alabama.
Conflicts of interestThe authors declare to have no conflicts of interest.
The authors are grateful to the Child Health Foundation in Birmingham, Alabama for their grant support for this work, to the participating patients and families, and to Dr. Silvia Castillo and cytogeneticist Ana María Fuentes for review of the conventional karyotype results.
Please cite this article as: Lay-Son G, Espinoza K, Vial C, Rivera JC, Guzmán ML, Repetto GM. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J Pediatr (Rio J). 2015;91:189–95.




