



Distal renal tubular acidosis (dRTA) is characterized by metabolic acidosis due to impaired renal acid excretion. The aim of this study was to demonstrate the genetic diagnosis of four children with dRTA through use of whole-exome sequencing.
MethodsTwo unrelated families were selected; a total of four children with dRTA and their parents, in order to perform whole-exome sequencing. Hearing was preserved in both children from the first family, but not in the second, wherein a twin pair had severe deafness. Whole-exome sequencing was performed in two pooled samples and findings were confirmed with Sanger sequencing method.
ResultsTwo mutations were identified in the ATP6V0A4 and ATP6V1B1 genes. In the first family, a novel mutation in the exon 13 of the ATP6V0A4 gene with a single nucleotide change GAC→TAC (c.1232G>T) was found, which caused a substitution of aspartic acid to tyrosine in position 411. In the second family, a homozygous recurrent mutation with one base-pair insertion (c.1149_1155insC) in exon 12 of the ATP6V1B1 gene was detected.
ConclusionThese results confirm the value of whole-exome sequencing for the study of rare and complex genetic nephropathies, allowing the identification of novel and recurrent mutations. Furthermore, for the first time the application of this molecular method in renal tubular diseases has been clearly demonstrated.
A acidose tubular renal distal (ATRd) é caracterizada por acidose metabólica devido a excreção renal de ácido prejudicada. O objetivo deste artigo é apresentar o diagnóstico genético de quatro crianças com ATRd utilizando o sequenciamento total do exoma.
MétodosSelecionamos duas famílias não relacionadas, totalizando quatro crianças com ATRd e seus pais, para realizar o sequenciamento total do exoma. A audição foi preservada em ambas as crianças da família um, porém em nenhuma criança da família dois, na qual um par de gêmeas teve perda auditiva severa. Realizamos o sequenciamento total do exoma em dois conjuntos de amostras e confirmamos os achados com o método de Sequenciamento de Sanger.
ResultadosDuas mutações foram identificadas nos genes ATP6V0A4 e ATP6V1B1. Na família um, detectamos uma nova mutação no éxon 13 do gene ATP6V0A4 com uma alteração em um nucleotídeo único GAC → TAC (c.1232G>T) que causou substituição de ácido aspártico por tirosina na posição 411. Na família dois, detectamos uma mutação recorrente do homozigoto com inserção de um par de bases (c.1149_1155insC) no éxon 12 do gene ATP6V1B1.
ConclusãoNossos resultados confirmam o valor do sequenciamento total do exoma para o estudo de nefropatias genéticas complexas, permitindo a identificação de mutações novas e recorrentes. Adicionalmente, demonstramos claramente pela primeira vez a aplicação desse método molecular em doenças tubulares renais.
Distal renal tubular acidosis (dRTA) is a rare and complex renal disease due to a defect in the excretion of acid load (H+ and ammonium ions) in alpha-intercalated cells of the collecting duct. The acid load accumulation in the distal nephron results in consumption and reduction of the bicarbonate/CO2 buffer in blood.1 The main clinical features of dRTA are vomiting, diarrhea, and/or constipation, loss of appetite, polydipsia, and polyuria. Chronic acidosis and secondary alterations such as vomiting, polyuria, and dehydration affect growth, leading to failure to thrive. Ultrasound studies can show nephrocalcinosis and/or nephrolithiasis.2 In general, dRTA has good prognosis if it is diagnosed at an early age and alkaline treatment is continued. Untreated, dRTA causes growth retardation and rickets in children and osteomalacia in adults. Deterioration of renal function can occur over the years.3
Distal RTA can be transmitted as either an autosomal dominant or an autosomal recessive trait.4 The autosomal dominant phenotype typically courses mildly in adolescence or adulthood;4 one parent suffers from and is the carrier of the disease, or it is due to de novo mutation. Mutations in the SLC4A1 gene in families with autosomal dominant dRTA have been identified.2,5,6 The symptoms in the autosomal recessive phenotype predominantly appear at infancy or early childhood, in which growth retardation is very common. This variant can occur with or without deafness, and parents are not affected.2 Autosomal recessive dRTA is associated with mutations in any of the following genes: SLC4A1,7ATP6V0A4, and ATP6V1B1.2,8 Individuals without hearing defects usually carry mutations in the ATP6V0A4 gene, while those with deafness have ATP6V1B1 gene mutations. In approximately 20% of the patients with dRTA, no mutations were found in any of these related genes.3 Indeed, there are dRTA patients with deafness without ATP6V1B1 gene mutations, and others with normal hearing who do not have ATP6V0A4 gene mutations.3 These findings suggest that other transporters or channels might cause dRTA. In terms of complexity, it is known that some patients with mutations in the ATP6V0A4 gene develop deafness only in the second decade of life. Thus, there remains much to be elucidated in terms of phenotype–genotype correlations.8–10 So far, more than 20 mutations in ATP6V0A4 are already known.
Whole-exome sequencing provides coverage of more than 95% of the exons, which contain 85% of disease-causing mutations in Mendelian disorders and many disease-predisposing single nucleotide polymorphisms (SNPs) throughout the genome.11,12 Whole-exome sequencing is worthwhile to evaluate the disease pathogenesis and to recognize new pathogenic genes or mutations associated to disorder, especially in Mendelian disorders.11,12 In this regard, the present study aimed to evaluate the usefulness of whole-exome sequencing for genetic diagnosis of dRTA.
Patients and methodsSubjects and clinical assessmentFour children with confirmed dRTA from two unrelated families were selected for this study. All patients were followed up at the Pediatric Nephrology Unit of the Federal University of Minas Gerais (UFMG), Brazil. The first family (Family 1) consisted of two affected siblings, a girl and a boy, with dRTA but without deafness, and their unaffected parents. The second family (Family 2) had a monozygotic twin pair (two girls), diagnosed with dRTA and nerve deafness, and their healthy mother; the father is unknown. All patients were submitted to a systematic protocol including clinical and nutritional evaluation, laboratory measurements, renal ultrasonography, and genetic analysis. Informed consent, approved by the institutional Ethics Board of UFMG, was obtained from all participants; in the case of children, it was also obtained from their parent/guardian.
DNA extractionGenomic DNA was extracted from 5mL of dRTA patients and their parent's peripheral blood, using a QIAamp Blood DNA mini Kit (Qiagen®, Milano, Italy) according to the manufacturer's instructions. All samples were quality controlled for purity using a NanoDrop spectrophotometer (Thermo Scientific®, Waltham, USA). DNA samples were stored at −20°C until usage.
Whole-exome sequencingExome sequencing was performed on two pools of samples to optimize the results. Samples were pooled considering the clinical features of the patients. The first pool had DNA from the two siblings with dRTA without deafness, and the second, from the twin sisters with dRTA associated with deafness. Array capture was used to isolate the relevant human genes (SeqCap EZ Human Exome Library v2.0, Roche®, Basel, Switzerland) and these genes were sequenced on the Illumina HiSeq 2000plataform (Sigma–Aldrich Corporation®, Missouri, USA).
Filtering dataThe following principle steps were taken to prioritize the high-quality variants: (i) variants within intergenic, intronic, and untranslated region (UTR) regions and synonymous mutations were excluded from downstream analysis; (ii) variants with quality score less than 20 were excluded; (iii) only the conservation score (phyloP) from comparison of human and 43 vertebrates higher than 3 were considered; (iv) after this prior selection, the remaining genes were filtered by the function. The software PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) predicted possible impact of variants. The final set of selected variants was visually inspected using an Integrative Genomics Viewer.13 Previously described polymorphic variants in public data were investigated and compared with the variations found in the current exome. The selected mutations to be investigated in each group of this study were not found in previous exome sequences (http://evs.gs.washington.edu/EVS/).
Validation of dataPolymerase chain reaction (PCR) Sanger sequencing was used in the analysis to confirm the data. All patients and their parents were submitted to the PCR. Amplification products of appropriate size were identified using polyacrylamide gel electrophoresis. Products were purified using the QIAquick PCR purification kit (Qiagen®, Milano, Italy) and then submitted to sequencing reaction using both forward and reverse primers with the ABI BigDye Terminator Cycle Sequencing Kit v. 3.1 on an ABI PRISM 3730XL Genetic Analyzer (Applied Biosystems®, Foster City, USA). Each read was aligned to the reference sequence, and mutations were identified with Sequencer software (http://www.genecodes.com). All primers were designed using the online tool Primer3. The primers for exon 12 of ATP6V1B1 gene were: 5′TTGACCCCTCGGAATGTAGG3′ and 5′CCGGACCCTCTTCTCCTTAC3′ (product size of 238 base pairs). The primers for exon 13 of ATP6V1B1 were: 5′ATGCAAATCGTGGAGCTGTG3′ and 5′ATGAATCAGGGCAAGACGGT3′ (product size of 264 base pairs).
Structural studies of mutationsProtein and DNA sequence alignments were performed using the ClustalW and the MultAlin (http://multalin.toulouse.inra.fr/multalin/), respectively. The prediction of amino acid substitution on the biological function of the protein was evaluated using both PolyPhen-2 and Provean software (http://provean.jcvi.org and http://genetics.bwh.harvard.edu/pph2/, respectively).
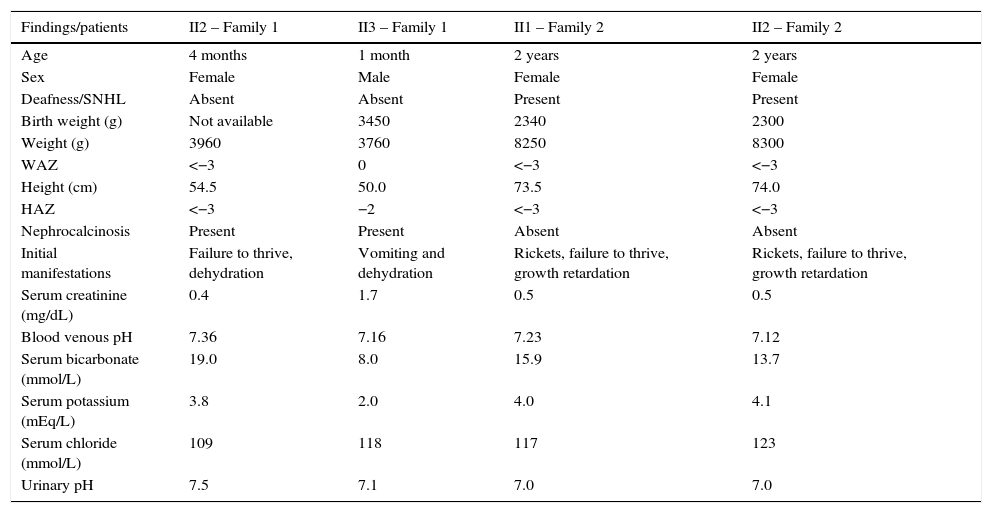
ResultsdRTA without deafnessThis family consisted of two siblings, a 13-year-old girl and her 7-year-old brother, with well-defined dRTA. The girl was the proband, diagnosed with dRTA at the age of 4 months. The initial findings were failure to thrive, hyperchloremic metabolic acidosis with abnormally high urine pH (7.0), normal venous blood pH (7.36), normal glomerular filtration rate, and nephrocalciosis. The boy was diagnosed in his 1st month of life, after a severe dehydration with metabolic acidosis, hypokalemia, transient elevation of serum creatinine, and hypocalcemia. His first renal ultrasonography showed bilateral nephrocalcinosis. Table 1 summarizes clinical and biochemical manifestations at baseline leading to the diagnosis of dRTA in each patient. The parents were unaffected, and had an older child that died by the age of 4 months with similar symptoms.
Clinical and biochemical findings at baseline of patients with dRTA.
| Findings/patients | II2 – Family 1 | II3 – Family 1 | II1 – Family 2 | II2 – Family 2 |
|---|---|---|---|---|
| Age | 4 months | 1 month | 2 years | 2 years |
| Sex | Female | Male | Female | Female |
| Deafness/SNHL | Absent | Absent | Present | Present |
| Birth weight (g) | Not available | 3450 | 2340 | 2300 |
| Weight (g) | 3960 | 3760 | 8250 | 8300 |
| WAZ | <−3 | 0 | <−3 | <−3 |
| Height (cm) | 54.5 | 50.0 | 73.5 | 74.0 |
| HAZ | <−3 | −2 | <−3 | <−3 |
| Nephrocalcinosis | Present | Present | Absent | Absent |
| Initial manifestations | Failure to thrive, dehydration | Vomiting and dehydration | Rickets, failure to thrive, growth retardation | Rickets, failure to thrive, growth retardation |
| Serum creatinine (mg/dL) | 0.4 | 1.7 | 0.5 | 0.5 |
| Blood venous pH | 7.36 | 7.16 | 7.23 | 7.12 |
| Serum bicarbonate (mmol/L) | 19.0 | 8.0 | 15.9 | 13.7 |
| Serum potassium (mEq/L) | 3.8 | 2.0 | 4.0 | 4.1 |
| Serum chloride (mmol/L) | 109 | 118 | 117 | 123 |
| Urinary pH | 7.5 | 7.1 | 7.0 | 7.0 |
Roman numerals indicate the family position on the pedigree: II2, Family 1 refers to the second proband (daughter) of family one; II3, Family 1 refers to the third proband (son) of family one; II1, Family 2 – the first twin daughter of family two; II2, Family 2 – the second twin daughter; SNHL, sensorineural hearing loss; WAZ, weight-for-age z-score; HAZ, height-for-age z-score.
Whole-exome sequencing conducted in this group generated 3577 single nucleotide variations (SNVs) and 416 small insertions and deletions (INDELs). Filtering for variants was applied to select the candidate gene (Table 2).
Variant prioritizing for Family 1 and for Family 2.
| Parameters | Family 1 | Family 2 |
|---|---|---|
| Number of variants | Number of variants | |
| Total variants | 3993 | 6791 |
| Intergenic, intronic, and UTR regions and synonymous mutations were excluded | 1445 | 1912 |
| Quality score <20 were excluded | 879 | 1012 |
| PhyloP <3 were excluded | 131 | 215 |
| Selection by function | 15 | 14 |
| PolyPhen <0.7 were excluded | 10 | 6 |
| Selection using cross-reference gene database and IGV | 1 | 1 |
| Candidate gene | 1 | 1 |
UTR, untranslated region.
After filtering the exome data, the ATP6V0A4 gene was selected for study. A single nucleotide change GAC→TAC (c.1232G>T) in exon 13 was observed, which caused an amino acid substitution: aspartic acid to tyrosine in position 411 (p.D411Y). This amino acid change was predicted to be damaging by Provean and PolyPhen-2. This mutation occurs at an evolutionarily conserved amino acid and affects highly preserved residues (data not shown).
The patients and their parents were submitted to Sanger sequencing by using the designed primer for exon 13 of the ATP6V0A4 gene. The two siblings presented the same mutation in homozygosis (c.1232G>T), while both parents had a heterozygous trace (Fig. 1A and B). This is a novel autosomal recessive dRTA mutation.
 The pedigree shows the affected statuses, individual identifiers, and genotypes at codon 411. The arrow indicates the proband. (B) DNA sequence chromatograms in which the two affected siblings have homozygous G to T substitution at c.1232. This substitution occurs in heterozygosis in both parents. WT, wild type. *Mutated nucleotide.")
Identification, pedigree of Family 1, and results of sequencing for c.1232G>T mutation. (A) The pedigree shows the affected statuses, individual identifiers, and genotypes at codon 411. The arrow indicates the proband. (B) DNA sequence chromatograms in which the two affected siblings have homozygous G to T substitution at c.1232. This substitution occurs in heterozygosis in both parents. WT, wild type. *Mutated nucleotide.
This family consisted of a twin pair of girls with dRTA in association with nerve deafness. The girls were diagnosed at the age of 2 after a long period of treating for rickets and growth retardation with only nutritional support. Clinical and biochemical features at baseline are shown in Table 1.
Whole-exome sequencing conducted in the family two generated 4375 SNVs and 2416 INDELs. After filtering the variants, only one candidate gene remained (Table 2).
Based on the exome data, the ATP6V1B1 gene was selected as the candidate in this group. A homozygous one base-pair insertion (c.1149_1155insC) in exon 12 was detected (Fig. 2A). The two dRTA twin sisters presented the insertion described. PCR of their unaffected mother was performed (father unknown). The two siblings had the same homozygous mutation, while the mother presented this insertion in heterozygosis (Fig. 2B and C).
 Identification of the c.1149_1155insC mutation using MultAlin. (B) The pedigree shows the affection statuses, individual identifiers, and genotypes at c.1149-1155insC. The arrow indicates the probands and the individual with “?\" has uncertain genotype status. (C) Direct DNA sequence chromatograms of family members, in which the two affected siblings have homozygous insertion of a C and the mother has a heterozygous trace, as marked by the red box. WT, wild type.")
Identification, pedigree of Family 2, and results of sequencing for the c.1149_1155insC mutation. (A) Identification of the c.1149_1155insC mutation using MultAlin. (B) The pedigree shows the affection statuses, individual identifiers, and genotypes at c.1149-1155insC. The arrow indicates the probands and the individual with “?" has uncertain genotype status. (C) Direct DNA sequence chromatograms of family members, in which the two affected siblings have homozygous insertion of a C and the mother has a heterozygous trace, as marked by the red box. WT, wild type.
Several DNA errors are located in exons leading to structural alterations in proteins and functional changes.3 In this manner, whole-exome analyzes these exons in a rapid and cost-effective manner, allowing genetic evaluation of complex and monogenic diseases.14,15 In rare disorders, the use of whole-exome may minimize the failure in detecting mutations at hot-spots regions. Nonetheless, direct Sanger sequencing is still considered the most accurate method to find mutations, since other gene testing techniques, such as whole exome, may not detect all sequence variations. The search in a whole-genome basis and validation of findings with Sanger sequencing method appears to be an efficient way to determine genetic causality of a disorder. However, the incorporation of these next-generation technologies into clinical practice is still challenging, since these techniques are remain very expensive, and data interpretation is laborious and difficult.11,12 Recent studies suggest that whole-exome sequencing would be useful to evaluate disease pathogenesis and to recognize new pathogenic genes or mutations associated to disorder, special in Mendelian disorders.16 Thus, the present study used whole exome followed by Sanger sequencing as a strategy for genetic diagnosis of dRTA in four children.
These results showed, for the first time, the utility of whole-exome sequencing in renal tubular disorders, by allowing the identification of a recurrent and a new pathogenic mutation in dRTA. Inherited forms of dRTA have three variants: autosomal dominant and autosomal recessive, with or without deafness.4 Dominant disease typically presents more mildly in adolescence or adulthood, and it has been only associated to mutations in the bicarbonate/chloride exchanger (AE1). However, the recessive variant occurs in infancy or early childhood, in which growth retardation is very common,4 as observed in the present cases. Autosomal recessive dRTA has been associated with mutations in the genes ATP6V1B1 and ATP6V0A4, which encode the subunits a4 and B1 of the vacuolar-type proton ATPase (V- or H+-ATPase), respectively.17–22 In addition, mutations in the SLC4A1 gene, responsible for the expression of AE1 proteins, have been also detected in cases of autosomal recessive dRTA without deafness.23–28
Indeed, mutations in different subunits of the proton pump that are expressed in kidney and ear tissues can cause tubular defects associated with deafness.17 The vacuolar-type proton ATPase (V- or H+-ATPase) is a pump with multiple subunits that is essential for normal acidification. Two structural domains form this pump: membrane-bound V0 and cytoplasmic or peripheral V1. Each domain comprises multiple subunits (a–e and A–H, respectively), which are responsible for ATP hydrolysis and proton transport, respectively.4 The ATP6V1B1 gene encodes the B1 subunit, while the ATP6V0A4 gene encodes the a4 subunit. The vacuolar-type proton ATPase is expressed apically on renal α-intercalated cells, and in the cochlea and endolymphatic sac. Based on the type of hearing loss, these types of mutations can be suspected. Conductive deafness was observed in mutations of the intracellular isoform of carbonic anidrase (CA), whereas sensorineural hearing loss (SNHL) has been associated with ATP6V1B1 and ATP6V0A4 mutations.18–20 Accordingly, a homozygous one base-pair insertion (c.1149_1155insC) was found in exon 12 of the ATP6V1B1 gene in twin sisters with SNHL. Mutations in the SLC4A1 gene usually do not have any association with deafness.22–28 Therefore, the presence and the kind of deafness are helpful in distinguishing different forms of dRTA.
In the two families of this study, parents were unaffected and dRTA had early onset, leading to growth impairment during infancy. Therefore, mutations in the SLC4A1 gene (AE1 proteins) were highly unlikely in the patients. In Family 1, exome sequencing identified a novel homozygous mutation in the ATP6V0A4 gene. Based on previous reports10 and on the clinical and biochemical features, this gene was selected as a potential candidate to search for mutations, since no hearing loss was detected in the affected patients. A single nucleotide change in exon 13 was observed that caused an amino acid substitution: aspartic acid to tyrosine in position 411. This aminoacid change was predicted to be damaging by Provean and PolyPhen-2. Unfortunately, no functional studies were performed to decipher the precise role of this mutation. However, it should be mentioned that this mutation occurs at an evolutionarily conserved aminoacid and affects highly preserved residues. In addition, the substitution of aspartic acid by tyrosine may change chemical properties of the protein at critical regions. For instance, this change may alter the isoelectric point of the protein, considering that tyrosine is a neutral aminoacid, while aspartic acid is an acid.
The twin girls of family two had SNHL and early-onset symptoms of dRTA. The phenotypes, together with the whole-exome data, led the authors to investigate the ATP6V1B1 gene. Accordingly, a previously described mutation was found,9 which was also confirmed by Sanger sequencing. It should be pointed, however, that ATP6V1B1 or ATP6V0A4 gene mutations have not been not found in some families with primary recessive forms of dRTA. There are numerous other candidate genes for recessive dRTA,29 especially those genes related to the proton transporters. In this regard, the use of whole-exome sequencing together with the phenotype characteristics may result in the discovery of new mutations and genetic alterations in this complex and rare disease.
In summary, whole-exome sequencing followed by Sanger sequencing was a successful strategy in identifying novel and recurrent mutations in these cases of dRTA. However, which genetic variants are potentially causative of renal tubular transport alterations remains to be elucidated, especially in recessive forms dRTA.
Conflicts of interestThe authors declare no conflicts of interest.
This study was partially supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil) and FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Brazil), by the Grant INCT-MM (Instituto Nacional de Ciência e Tecnologia – Medicina Molecular: FAPEMIG: CBB-APQ-00075-09/CNPq 573646/2008-2). Dr. LA De Marco, Dr. EA Oliveira, Dr. DM Miranda, and Dr. AC Simões e Silva received a research grant from CNPq.
Please cite this article as: Pereira PC, Melo FM, De Marco LA, Oliveira EA, Miranda DM, Simões e Silva AC. Whole-exome sequencing as a diagnostic tool for distal renal tubular acidosis. J Pediatr (Rio J). 2015;91:583–9.




