



Distal renal tubular acidosis (dRTA) is characterized by metabolic acidosis due to impaired renal acid excretion. The aim of this study was to demonstrate the genetic diagnosis of four children with dRTA through use of whole‐exome sequencing.
MethodsTwo unrelated families were selected; a total of four children with dRTA and their parents, in order to perform whole‐exome sequencing. Hearing was preserved in both children from the first family, but not in the second, wherein a twin pair had severe deafness. Whole‐exome sequencing was performed in two pooled samples and findings were confirmed with Sanger sequencing method.
ResultsTwo mutations were identified in the ATP6V0A4 and ATP6V1B1 genes. In the first family, a novel mutation in the exon 13 of the ATP6V0A4 gene with a single nucleotide change GAC→TAC (c.1232G>T) was found, which caused a substitution of aspartic acid to tyrosine in position 411. In the second family, a homozygous recurrent mutation with one base‐pair insertion (c.1149_1155insC) in exon 12 of the ATP6V1B1 gene was detected.
ConclusionThese results confirm the value of whole‐exome sequencing for the study of rare and complex genetic nephropathies, allowing the identification of novel and recurrent mutations. Furthermore, for the first time the application of this molecular method in renal tubular diseases has been clearly demonstrated.
A acidose tubular renal distal (ATRd) é caracterizada por acidose metabólica devido à excreção renal de ácido prejudicada. O objetivo deste artigo é apresentar o diagnóstico genético de quatro crianças com ATRd com uso do sequenciamento total do exoma.
MétodosSelecionamos duas famílias não relacionadas, quatro crianças com ATRd e seus pais, para fazer o sequenciamento total do exoma. A audição foi preservada em ambas as crianças da família um, porém em nenhuma criança da família dois, na qual um par de gêmeas teve perda auditiva severa. Fizemos o sequenciamento total do exoma em dois conjuntos de amostras e confirmamos os achados com o método de sequenciamento de Sanger.
ResultadosDuas mutações foram identificadas nos genes ATP6V0A4 e ATP6V1B1. Na família um, detectamos uma nova mutação no éxon 13 do gene ATP6V0A4 com uma alteração em um nucleotídeo único GAC → TAC (c.1232G>T) que causou substituição de ácido aspártico por tirosina na posição 411. Na família dois, detectamos uma mutação recorrente do homozigoto com inserção de um par de bases (c.1149_1155insC) no éxon 12 do gene ATP6V1B1.
ConclusãoNossos resultados confirmam o valor do sequenciamento total do exoma para o estudo de nefropatias genéticas complexas e permitem a identificação de mutações novas e recorrentes. Adicionalmente, demonstramos claramente pela primeira vez a aplicação desse método molecular em doenças tubulares renais.
A acidose tubular renal distal (ATRd) é uma doença renal rara e complexa devido a um defeito na excreção da carga de ácidos (H+ e íons de amônia) em células alfa intercaladas do ducto coletor. O acúmulo da carga de ácidos no néfron distal resulta no consumo e na redução do tampão de bicarbonato/CO2 no sangue.1 As principais características clínicas da ATRd são vômito, diarreia e/ou constipação, perda de apetite, polidipsia e poliúria. A acidose crônica e as alterações secundárias como vômito, poliúria e desidratação afetam o crescimento e levam a um déficit de crescimento. Estudos de ultrassom podem mostrar nefrocalcinose e/ou nefrolitíase.2 Em geral, a ATRd apresenta bom prognóstico caso diagnosticada precocemente e o tratamento alcalino é contínuo. Caso não tratada, a ATRd causa retardo do crescimento e raquitismo em crianças e osteomalacia em adultos. Pode ocorrer deterioração da função renal ao longo dos anos.3
A ATR distal pode ser transmitida como uma característica autossômica dominante ou recessiva.4 O fenótipo autossômico dominante normalmente aparece moderadamente na adolescência ou na vida adulta;4 um dos pais padece e é o portador da doença ou a doença se dá por meio de uma mutação de novo. Foram identificadas mutações no gene SLC4A1 em famílias com ATRd autossômica dominante.2,5,6 Os sintomas no fenótipo autossômico recessivo aparecem predominantemente na infância ou na primeira infância, quando o retardo do crescimento é muito comum. Essa variável pode ocorrer com ou sem surdez e os pais não são afetados.2 A ATRd autossômica recessiva está associada a mutações em quaisquer dos seguintes genes: SLC4A1,7ATP6V0A4 e ATP6V1B1.2,8 Indivíduos sem deficiências auditivas normalmente são portadores de mutações no gene ATP6V0A4, ao passo que aqueles com surdez apresentam mutações no gene ATP6V1B1. Em aproximadamente 20% dos pacientes com ATRd, nenhuma mutação foi encontrada em quaisquer desses genes relacionados.3 De fato, existem pacientes com ATRd com surdez sem mutações no gene ATP6V1B1 e outros com audição normal que não apresentam mutações no gene ATP6V0A4.3 Esses achados sugerem que outros transportadores ou canais podem causar ATRd. Em termos de complexidade, sabe‐se que alguns pacientes com mutações no gene ATP6V0A4 desenvolvem surdez apenas na segunda década de vida. Portanto, há muitos fatores a serem elucidados em termos de correlações fenótipo‐genótipo.8–10 Até agora, já são conhecidas mais de 20 mutações no gene ATP6V0A4.
O sequenciamento total do exoma fornece cobertura de mais de 95% dos éxons, que contêm 85% das mutações causadoras de doenças em doenças mendelianas e muitos polimorfismos de nucleotídeo simples (SNPs) com predisposição para doenças em todo o genoma.11,12 O sequenciamento total do exoma é interessante para avaliar a patogênese da doença e reconhecer novos genes ou mutações patogênicos relacionados a doenças, principalmente às doenças mendelianas.11,12 Nesse sentido, este estudo pretendeu avaliar a utilidade do sequenciamento total do exoma para o diagnóstico genético de ATRd.
Pacientes e métodosAvaliação individual e clínicaQuatro crianças com ATRd confirmada de duas famílias não relacionadas foram selecionadas para este estudo. Todos os pacientes foram acompanhados na Unidade de Nefrologia Pediátrica da Universidade Federal de Minas Gerais (UFMG), Brasil. A primeira família (família um) consistia em dois irmãos afetados, uma menina e um menino, com ATRd, porém sem surdez, com pais não afetados. A segunda família (família dois) apresentava um par de gêmeas monozigóticas (duas meninas), diagnosticadas com ATRd e surdez nervosa, com mãe saudável; o pai é desconhecido. Todos os pacientes foram submetidos a um protocolo sistemático, incluindo avaliação clínica e nutricional, medições laboratoriais, ultrassonografia renal e análise genética. O consentimento informado, aprovado pelo conselho de ética institucional da UFMG, foi obtido de todos os participantes; no caso das crianças, também foi obtido de seus pais e/ou responsáveis legais.
Extração do DNAO DNA genômico foi extraído de 5mL de sangue periférico de pacientes com ATRd e de seus pais, com o uso do minikit Qiamp Blood DNA (Qiagen®, Milão, Itália), de acordo com as instruções do fabricante. Todas as amostras tiveram o controle de qualidade verificado com relação à pureza com o uso de um espectrofotômetro Nanodrop (Thermo Scientific®, Waltham, EUA). As amostras de DNA foram armazenadas a −20°C até o uso.
Sequenciamento total do exomaO sequenciamento do exoma foi feito em dois conjuntos de amostras para aprimorar os resultados. As amostras foram divididas em conjuntos no que diz respeito às características clínicas dos pacientes. O primeiro conjunto apresentava DNA dos dois irmãos com ATRd sem surdez e o segundo, das irmãs gêmeas com ATRd relacionada à surdez. A captação do array foi usada para isolar os respectivos genes humanos (SeqCap EZ Human Exome Library v2.0, Roche®, Basileia, Suíça) e esses genes foram sequenciados na plataforma Illumina HiSeq 2000 (Sigma‐Aldrich Corporation®, Missouri, EUA).
Dados de filtragemAs principais etapas a seguir foram feitas para priorizar as variáveis de alta qualidade: (i) variáveis em regiões intergenéticas, intrônicas e não traduzidas (UTR) e mutações sinônimas foram excluídas da análise a jusante; (ii) variáveis com índice de qualidade menor do que 20 foram excluídas; (iii) somente o índice de conservação (phyloP) da comparação de humanos e 43 vertebrados acima de 3 foi considerado; (iv) após essa seleção inicial, os genes restantes foram filtrados pela função. O software Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) previu possíveis impactos de variáveis. O conjunto final de variáveis selecionadas passou por inspeção visual com o uso do Integrative Genomics Viewer.13 Variáveis polimórficas anteriormente descritas em dados públicos foram investigadas e comparadas com as variações encontradas no exoma atual. As mutações selecionadas para investigação em cada grupo deste estudo não foram encontradas em sequenciamentos de exomas anteriores (http://evs.gs.washington.edu/EVS/).
Validação de dadosO sequenciamento de Sanger da reação em cadeia da polimerase (PCR) foi usado na análise para confirmar os dados. Todos os pacientes e seus pais foram submetidos a PCR. Produtos de amplificação de tamanho adequado foram identificados com o uso de eletroforese em gel de poliacrilamida. Os produtos foram purificados com o kit QIAquick PCR (Qiagen®, Milão, Itália) e então submetidos a uma reação de sequenciamento com o uso tanto de iniciadores forward quanto reverse com o ABI BigDye Terminator Cycle Sequencing Kit v3.1 em um analisador genético ABI PRISM 3730XL (Applied Biosystems®, Foster City, EUA). Cada leitura foi alinhada ao sequenciamento de referência e as mutações foram identificadas com o software Sequencher (http://www.genecodes.com). Todos os iniciadores foram projetados com a ferramenta on‐line Primer3. Os iniciadores do éxon 12 do gene ATP6V1B1 foram: 5’TTGACCCCTCGGAATGTAGG3’ e 5’CCGGACCCTCTTCTCCTTAC3’ (tamanho do produto: 238 pares de bases). Os iniciadores do éxon 13 do gene ATP6V1B1 foram: 5’ATGCAAATCGTGGAGCTGTG3’ e 5’ATGAATCAGGGCAAGACGGT3’ (tamanho do produto: 264 pares de bases).
Estudos estruturais das mutaçõesOs alinhamentos de proteínas e sequências de DNA foram feitos com os softwares ClustalW e MultAlin (http://multalin.toulouse.inra.fr/multalin/), respectivamente. A previsão da substituição de aminoácidos na função biológica da proteína foi avaliada com o uso tanto do software PolyPhen‐2 quanto do Provean (http://provean.jcvi.org e http://genetics.bwh.harvard.edu/pph2/, respectivamente).
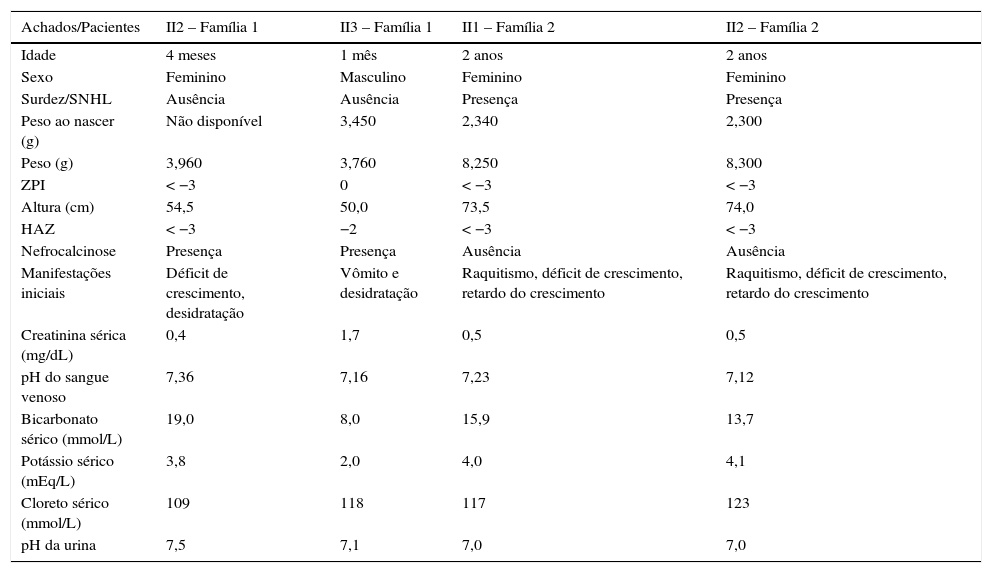
ResultadosATRd sem surdezEssa família consistia em dois irmãos, uma menina de 13 anos e seu irmão de sete, com uma ATRd bem definida. A menina foi a probanda, diagnosticada com ATRd aos quatro meses. Os achados iniciais foram déficit de crescimento, acidose metabólica hiperclorêmica com pH da urina excepcionalmente alto (7,0), pH do sangue venoso normal (7,36), taxa de filtração glomerular normal e nefrocalcinose. O menino foi diagnosticado no primeiro mês de vida, após uma desidratação grave com acidose metabólica, hipocalemia, elevação transitória da creatinina sérica e hipocalcemia. Sua primeira ultrassonografia renal mostrou nefrocalcinose bilateral. A tabela 1 resume as manifestações clínicas e bioquímicas básicas que levaram ao diagnóstico de ATRd em cada paciente. Os pais não foram afetados e tiveram um filho mais velho que faleceu aos quatro meses com sintomas semelhantes.
Achados clínicos e bioquímicos iniciais de pacientes com ATRd
| Achados/Pacientes | II2 – Família 1 | II3 – Família 1 | II1 – Família 2 | II2 – Família 2 |
|---|---|---|---|---|
| Idade | 4 meses | 1 mês | 2 anos | 2 anos |
| Sexo | Feminino | Masculino | Feminino | Feminino |
| Surdez/SNHL | Ausência | Ausência | Presença | Presença |
| Peso ao nascer (g) | Não disponível | 3,450 | 2,340 | 2,300 |
| Peso (g) | 3,960 | 3,760 | 8,250 | 8,300 |
| ZPI | < −3 | 0 | < −3 | < −3 |
| Altura (cm) | 54,5 | 50,0 | 73,5 | 74,0 |
| HAZ | < −3 | −2 | < −3 | < −3 |
| Nefrocalcinose | Presença | Presença | Ausência | Ausência |
| Manifestações iniciais | Déficit de crescimento, desidratação | Vômito e desidratação | Raquitismo, déficit de crescimento, retardo do crescimento | Raquitismo, déficit de crescimento, retardo do crescimento |
| Creatinina sérica (mg/dL) | 0,4 | 1,7 | 0,5 | 0,5 |
| pH do sangue venoso | 7,36 | 7,16 | 7,23 | 7,12 |
| Bicarbonato sérico (mmol/L) | 19,0 | 8,0 | 15,9 | 13,7 |
| Potássio sérico (mEq/L) | 3,8 | 2,0 | 4,0 | 4,1 |
| Cloreto sérico (mmol/L) | 109 | 118 | 117 | 123 |
| pH da urina | 7,5 | 7,1 | 7,0 | 7,0 |
Os números romanos indicam a posição da família na linhagem: II2, família 1 significa a segunda probanda (filha) da família um; II3, família 1 significa o terceiro probando (filho) da família um; II1, família 2 – a primeira filha gêmea da família dois; II2, família 2 – a segunda filha gêmea; SNHL, perda auditiva neurossensorial; ZPI, escores z de peso por idade; HAZ, escores z de altura por idade.
O sequenciamento total do exoma feito nesse grupo gerou 3577 variantes de nucleotídeo único (SNVs) e 416 pequenas inserções e deleções (INDELs). A filtragem de variáveis foi aplicada para selecionar o gene candidato (tabela 2).
Priorização da variável para a família um e família dois
| Família 1 | Família 2 | |
|---|---|---|
| Parâmetros | Número de variáveis | Número de variáveis |
| Total de variáveis | 3.993 | 6.791 |
| Regiões intergênicas, intrônicas e UTR e mutações sinônimas foram excluídas | 1.445 | 1.912 |
| Variáveis com índice de qualidade < 20 foram excluídas | 879 | 1.012 |
| PhyloP < 3 foram excluídas | 131 | 215 |
| Seleção por função | 15 | 14 |
| PhyloP<0,7 foram excluídas | 10 | 6 |
| Seleção com o uso da base de dados de genes com referência cruzada e o aplicativo IGV | 1 | 1 |
| Gene candidate | 1 | 1 |
UTR, região não traduzida.
Após filtrar os dados do exoma, selecionamos o gene ATP6V0A4 para estudo. Observamos uma alteração em um nucleotídeo único GAC → TAC (c.1232G>T) no éxon 13 que causou substituição de um aminoácido: ácido aspártico por tirosina na posição 411 (p.D411Y). Essa alteração no aminoácido foi preditiva de ser danosa pelo Provean e pelo PolyPhen‐2. Essa mutação ocorre em um aminoácido evolutivamente conservado e afeta resíduos altamente preservados (dados não apresentados).
Os pacientes e seus pais foram submetidos ao sequenciamento de Sanger ao usar o iniciador projetado para o éxon 13 do gene ATP6V0A4. Os dois irmãos apresentaram a mesma mutação em homozigose (c.1232G>T), ao passo que ambos os pais apresentaram um traço heterozigótico (fig. 1A e B). Essa é uma nova mutação autossômica recessiva de ATRd.
 A linhagem mostra os estados afetados, identificadores individuais e genótipos no códon 411. A seta indica o probando. B) Cromatogramas do sequenciamento do DNA em que os dois irmãos afetados têm substituição do homozigoto G por T em c.1232. Essa substituição ocorre em heterozigose em ambos os pais. WT, alelo selvagem. *Sequência nucleotídica mutada.")
Identificação, linhagem da família um e resultados de sequenciamento para mutação c.1232G>T. A) A linhagem mostra os estados afetados, identificadores individuais e genótipos no códon 411. A seta indica o probando. B) Cromatogramas do sequenciamento do DNA em que os dois irmãos afetados têm substituição do homozigoto G por T em c.1232. Essa substituição ocorre em heterozigose em ambos os pais. WT, alelo selvagem. *Sequência nucleotídica mutada.
Essa família consistia em um par de gêmeas com ATRd associada a surdez nervosa. As meninas foram diagnosticadas aos dois anos após um longo período de tratamento para raquitismo e retardo do crescimento somente com assistência nutricional. Características clínicas e bioquímicas no início são apresentadas na tabela 1.
O sequenciamento total do exoma conduzido na família dois gerou 4375 SNVs e 2416 INDELs. Após filtrar as variáveis, obtivemos somente um gene candidato restante (tabela 2).
Selecionamos, com base nos dados do exoma, o gene ATP6V1B1 como candidato nesse grupo. Uma inserção de um par de bases homozigótico (c.1149_1155insC) no exoma 12 foi detectada (fig. 2A). As duas gêmeas com ATRd apresentaram a inserção descrita. Fizemos a PCR da mãe não afetada (pai desconhecido). Os dois irmãos apresentaram a mesma mutação homozigótica, ao passo que a mãe apresentou essa inserção na heterozigose (fig. 2B e C).
 Identificação da mutação c.1149_1155insC com o software MultAlin. B) A linhagem mostra os estados afetados, identificadores individuais e genótipos em c.1149‐1155insC. A seta indica os probandos e o indivíduo com “?” tem situação do genótipo incerta. C) Cromatogramas de sequenciamento de DNA direto de membros da família em que os dois irmãos afetados têm inserção homozigótica de um C e a mãe tem um traço heterozigótico, conforme marcado pela caixa vermelha. WT, alelo selvagem.")
Identificação, linhagem da família dois e resultados de sequenciamento para mutação c.1149_1155insC. A) Identificação da mutação c.1149_1155insC com o software MultAlin. B) A linhagem mostra os estados afetados, identificadores individuais e genótipos em c.1149‐1155insC. A seta indica os probandos e o indivíduo com “?” tem situação do genótipo incerta. C) Cromatogramas de sequenciamento de DNA direto de membros da família em que os dois irmãos afetados têm inserção homozigótica de um C e a mãe tem um traço heterozigótico, conforme marcado pela caixa vermelha. WT, alelo selvagem.
Vários erros de DNA estão localizados em éxons e levam a alterações estruturais nas proteínas e alterações funcionais.3 Dessa forma, o sequenciamento total do exoma analisa esses éxons de uma maneira rápida e com bom custo‐benefício e permite avaliação genética das doenças complexas e monogenéticas.14,15 Em doenças raras, o uso do sequenciamento total do exoma pode minimizar a falha na detecção de mutações em regiões críticas. Por outro lado, o sequenciamento direto de Sanger ainda é considerado o método mais preciso para encontrar mutações, uma vez que outras técnicas de teste genético, como o sequenciamento total do exoma, poderão não detectar todas as variações da sequência. A busca em todo o genoma e a validação de achados com o método de sequenciamento Sanger parecem ser uma maneira eficiente de determinar a causalidade genética de uma doença. Entretanto, a forma como essas tecnologias de última geração serão incorporadas à prática clínica ainda é desafiadora, pois essas técnicas ainda são muito caras e a interpretação de dados é trabalhosa e difícil.11,12 Estudos recentes sugerem que o sequenciamento total do exoma seria útil para avaliar a patogênese da doença e reconhecer novos genes ou mutações patogênicos relacionados a doenças, principalmente às doenças mendelianas.16 Assim, no presente estudo, usamos o sequenciamento total do exoma seguido pelo sequenciamento de Sanger como uma estratégia para o diagnóstico genético de ATRd em quatro crianças.
Nossos resultados mostraram, pela primeira vez, a utilidade do sequenciamento total do exoma em doenças tubulares renais e permitiram a identificação de uma mutação recorrente e uma nova mutação patogênica na ATRd. As formas herdadas de ATRd apresentam três variáveis: autossômica dominante e autossômica recessiva, com ou sem surdez.4 A doença dominante normalmente se apresenta mais suavemente na adolescência ou na vida adulta e tem sido relacionada apenas a mutações no trocador bicarbonato/cloreto (AE1). Por outro lado, a variável recessiva ocorre na infância ou na primeira infância, quando o retardo do crescimento é muito comum,4 conforme observado em nossos casos. A ATRd autossômica recessiva tem sido relacionada a mutações nos genes ATP6V1B1 e ATP6V0A4, que codificam as subunidades a4 e B1da ATPase de próton do tipo vacuolar (V‐ ou H+‐ATPase), respectivamente.17‐22 Além disso, mutações no gene SLC4A1, responsável pela expressão de proteínas AE1, também foram detectadas em casos de ATRd autossômica recessiva sem surdez.23‐28
De fato, mutações em diferentes subunidades da bomba de próton expressas em tecidos renais e de ouvidos podem causar defeitos tubulares relacionados à surdez.17 A ATPase de próton do tipo vacuolar (V‐ ou H+‐ATPase) é uma bomba de várias subunidades essencial para a acidificação normal. Dois domínios estruturais formam essa bomba: V0 ligado à membrana e V1 citoplasmático ou periférico. Cada domínio incluía múltiplas subunidades (a–e e A–H, respectivamente), responsáveis pela hidrólise do ATP e transporte de prótons, respectivamente.4 O gene ATP6V1B1 codifica a subunidade B1, ao mesmo tempo em que o gene ATP6V0A4 codifica a subunidade a4. O próton ATPase vacuolar é expresso apicalmente em células renais α intercaladas, na cóclea e no saco endolinfático. Com base no tipo de perda auditiva, o tipo de mutações pode ser suspeito. A perda auditiva condutiva foi observada em mutações da isoforma intracelular de anidrase carbônica (AC), considerando que a perda auditiva neurossensorial (SNHL) foi associada a mutações nos genes ATP6V1B1 e ATP6V0A4.18‐20 Dessa forma, encontramos uma inserção de um par de bases homozigótico (c.1149_1155insC) no éxon 12 do gene ATP6V1B1 em gêmeas com SNHL. Mutações no gene SLC4A1 normalmente não apresentam associação com surdez.22‐28 Portanto, a presença e o tipo de surdez auxiliam na distinção de diferentes formas de ATRd.
Nas duas famílias deste estudo, os pais não foram afetados, a ATRd teve início precoce e levou ao impedimento do crescimento durante a infância. Portanto, as mutações no gene SLC4A1 (proteínas AE1) foram altamente diferentes em nossos pacientes. Na família um, o sequenciamento do exoma identificou uma nova mutação homozigótica no gene ATP6V0A4. Com base em relatórios anteriores10 e em nossas características clínicas e bioquímicas, esse gene foi selecionado como possível candidato para buscar mutações, pois nenhuma perda auditiva foi detectada nos pacientes afetados. Observamos uma única alteração nucleotídica no éxon 13 que causou substituição de um aminoácido: ácido aspártico por tirosina na posição 411. Essa alteração no aminoácido foi preditiva de ser danosa pelo Provean e pelo PolyPhen‐2. Infelizmente, não fizemos estudos funcionais para decifrar a função precisa dessa mutação. Entretanto, deve‐se mencionar que essa mutação ocorre em um aminoácido conservado evolutivamente e afeta resíduos altamente preservados. Além disso, a substituição de ácido aspártico por tirosina pode alterar as propriedades químicas da proteína em regiões críticas. Por exemplo, essa alteração pode modificar o ponto isoelétrico da proteína, considerando que a tirosina é um aminoácido neutro, ao passo que o ácido aspártico é um ácido.
As gêmeas da família dois apresentaram perda auditiva neurossensorial e sintomas de início precoce de ATRd. Os fenótipos juntamente com os dados do exoma total nos levaram a investigar o gene ATP6V1B1. Dessa forma, constatamos uma mutação previamente descrita,9 que também foi confirmada pelo sequenciamento de Sanger. Entretanto, deve‐se destacar que as mutações dos genes ATP6V1B1 ou ATP6V0A4 não foram encontradas em algumas famílias com formas recessivas primárias de ATRd. Há vários outros genes candidatos para a ATRd recessiva,29 principalmente os relacionados aos transportadores de prótons. Nesse sentido, o uso do sequenciamento total do exoma, juntamente com as características fenotípicas, pode resultar na descoberta de novas mutações e alterações genéticas nessa doença complexa e rara.
Em resumo, o sequenciamento total do exoma seguido pelo sequenciamento Sanger foi uma estratégia bem‐sucedida na identificação de novas e recorrentes mutações em nossos casos de ATRd. Entretanto, as variações genéticas possíveis causadoras de alterações no transporte tubular renal, principalmente nas formas recessivas de ATRd, ainda devem ser elucidadas.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Este estudo foi parcialmente patrocinado pelo Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brasil, e pela Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig), Brasil, através da Concessão do Instituto Nacional de Ciência e Tecnologia – Medicina Molecular (INCT‐MM): Fapemig: CBB‐APQ‐00075‐09/CNPq 573646/2008‐2. Dr. LA De Marco, Dr. EA Oliveira, Dr. DM Miranda e Dr. AC Simões e Silva receberam uma bolsa de pesquisa do CNPq.
Como citar este artigo: Pereira PC, Melo FM, De Marco LA, Oliveira EA, Miranda DM, Simões e Silva AC. Whole‐exome sequencing as a diagnostic tool for distal renal tubular acidosis. J Pediatr (Rio J). 2015;91:583–89.




