

The purpose of this study was to illustrate the association between vascular endothelial growth factor level and pulmonary artery hypertension in children with β-thalassemia major.
MethodThis case–control study was conducted on 116 children with β-thalassemia major; 58 of them had pulmonary artery hypertension. They were compared to 58 healthy children who were age and sex-matched (control group). Serum levels of vascular endothelial growth factor and echocardiographic assessment were done for all children.
ResultsVascular endothelial growth factor serum level was significantly higher in children with β-thalassemia major with pulmonary artery hypertension than in those without pulmonary artery hypertension, as well as in control groups (p<0.001). Vascular endothelial growth factor serum level had a significant positive correlation with pulmonary artery pressure and serum ferritin, as well as a significant negative correlation with the duration of chelation therapy. Logistic regression analysis revealed that elevated vascular endothelial growth factor (Odd Ratio=1.5; 95% Confidence Interval, 1.137–2.065; p=0.005) was an independent risk factor of pulmonary artery hypertension in such children. Vascular endothelial growth factor serum level at a cutoff point of >169pg/mL had 93.1% sensitivity and 93.1% specificity for the presence of pulmonary artery hypertension in children with β-thalassemia major.
ConclusionElevated vascular endothelial growth factor serum level is associated with pulmonary artery hypertension in children with β-thalassemia.
A finalidade deste estudo foi exemplificar a associação entre o nível de fator de crescimento endotelial vascular e a hipertensão arterial pulmonar em crianças com talassemia beta maior.
MétodoEste estudo caso-controle foi realizado em 116 crianças com talassemia beta maior; 58 das quais apresentaram hipertensão arterial pulmonar em comparação com 58 crianças saudáveis pareadas por idade e sexo (grupo de controle). Os níveis séricos do fator de crescimento endotelial vascular e a avaliação ecocardiográfica foram realizados em todas as crianças.
ResultadosO nível sérico do fator de crescimento endotelial vascular foi significativamente maior em crianças com talassemia beta maior com hipertensão arterial pulmonar que as crianças sem hipertensão arterial pulmonar e os grupos de controle (p<0,001). O nível sérico do fator de crescimento endotelial vascular apresentou uma correlação positiva significativa com a pressão arterial pulmonar e a ferritina sérica e correlação negativa significativa com a duração da terapia de quelação. A análise de regressão logística revelou que o fator de crescimento endotelial vascular elevado (RC=1,5; IC de 95%: 1,137-2,065; p=0,005) foi um fator de risco independente de hipertensão arterial pulmonar nessas crianças. O nível sérico do fator de crescimento endotelial vascular no ponto de corte> 169 (pg/mL) apresentou 93,1% de sensibilidade e 93,1% de especificidade na presença de hipertensão arterial pulmonar em crianças com talassemia beta maior.
ConclusãoO nível sérico do fator de crescimento endotelial vascular elevado está associado à hipertensão arterial pulmonar em crianças com talassemia beta.
Myocardial dysfunction and heart failure were considered as common causes of morbidity and mortality in children with β-thalassemia major, which were related mainly to chronic hemolysis and iron overload.1 However, pulmonary arterial hypertension (PAH) has emerged as a major risk factor for impaired right ventricular function in such patients.1 PAH is a multifactorial, complex vascular disorder. The main underlying mechanism of PAH in patients with β-thalassemia is not yet understood. Several interacting factors were suggested including platelet activation, chronic hemolysis, dysfunctional angiogenesis, oxidative stress, and iron overload.2 The process of chronic hemolysis in thalassemia causes an increased production of free hemoglobin, which eliminates nitric oxide that protects against vascular endothelial damage and releases the arginase enzyme into the circulation, which converts l-arginine to ornithine, thus bypassing nitric oxide production that also mediates vascular relaxation.3 PAH is a proangiogenic disease; however, the role of angiogenic factors such as the vascular endothelial growth factor (VEGF) in the pathogenesis of PAH is not well demonstrated.
Conventional echocardiography is a reliable, non-invasive tool for detection and follow-up of pulmonary arterial pressure; however, cardiac catheterization is the gold standard method for diagnosis of PAH.4 In addition to these traditional diagnostic methods, serum biomarkers related either to myocardial strain or to the underlying pathology are widely employed.5 As PAH is a progressive, irreversible, life-threatening disorder there is an urgent need for an easy, available, subjective tool to assess the individualized risk and progression of PAH in children with β-thalassemia major.
The pulmonary vascular endothelial cell is an important regulator of vascular tone. VEGF is a potent angiogenic peptide that regulates proliferation of vascular endothelium and vascular permeability, and it also promotes neovascularization.6 Elevated VEGF plasma level was suspected as one of the mechanisms involved in the vascular remodeling and development of PAH.7 VEGF upregulation was induced by exposure to hypoxia, as it is done in patients with chronic hemolytic anemia.8 Previous studies reported elevation of angiogenic cytokines in thalassemia such as VEGF, basic fibroblast growth factor, angiogenin, angiopoietin or VEGF and tumor necrosis factor.9
In an attempt to improve our understanding of the pathophysiological mechanism and risk factors of PAH in children with β-thalassemia major, we conducted this study to illustrate the association with VEGF serum level and PAH in those children. In addition, we explored the relationship between VEGF serum levels and the clinical data, chelation characteristics and serum ferritin in children with β-thalassemia major.
MethodsThis comparative case–control study was conducted on 116 children with a confirmed diagnosis of β-thalassemia major who received regular blood transfusion in the period from September 2015 to January 2017. Fifty-eight of them had pulmonary hypertension (mean pulmonary artery pressure>25mmHg) as confirmed by echocardiography, while the remaining 58 children had normal pulmonary artery pressure. Children who fulfilled our inclusion criteria were selected consecutively from the pediatric department of Al-Zahraa University hospital, Al-Azhar University, Cairo, Egypt. In addition, 58 healthy children (control group) that had no hematological disorders as confirmed by complete blood count and hemoglobin electrophoresis, did not have any cardiac abnormalities confirmed by echocardiography and fulfilled the same exclusion criteria for the patient group were selected. They were matched for age and sex with the thalassemia patients. Written informed consent was obtained from the children's guardians, and approval was provided by the local ethical committee from the faculty of medicine, Al-Azhar University, Cairo, Egypt.
Inclusion criteria included children with β-thalassemia major confirmed through hemoglobin electrophoresis; their ages ranged from 10 to 13 years. They received a regular blood transfusion every 1–2 months of an average amount of 150–250cc of packed RBCs at each transfusion. Children with congenital or rheumatic heart disease, heart failure, systemic chronic diseases other than thalassemia or any concomitant acute illness were excluded from the study.
History and clinical examinationDetailed history and thorough clinical examination were done to all children. Detailed data were recorded on demographics, the age of onset of the disease, the frequency of blood transfusion, the age of onset, the type of iron chelation therapy and history of splenectomy, as well as any symptoms of pulmonary congestion, low cardiac output or heart failure. The findings of systemic, abdominal and cardiac examinations were recorded in detail.
Serum levels of VEGF and echocardiographic assessment were done for all included children. We evaluated the association between serum levels of VEGF and PAH in children with β-thalassemia major and its relation to other clinical and laboratory data.
EchocardiographyAll patients and control children were referred for echocardiography using Philips HD7 XE system. The assessment was done using multiple transducers ranging from 3.5 to 7MHz with simultaneous electrocardiographic recording to allow timing of flow. M-mode and 2D echocardiography in both supine and left lateral position were used to estimate the pulmonary pressure and determine the diameter of cardiac chambers.
M-mode measurementsM-mode was carried out from the left ventricle parasternal short axis view by applying the cursor line perpendicular at mid-papillary level to estimate the following: left ventricular internal dimensions at the end of systole and diastole (LVESD, LVEDD) respectively, fractional shortening (FS%), ejection fraction (EF%), right ventricle (RV) diameter, posterior wall (PW) thickness, septal wall thickness, aortic diameter (AO) and right atrium (RA) diameter. EF% normal value varies from age range in 55–65% in children.
Two-dimensional echo (2D echo): 2D echo images were obtained from the parasternal (long and short axis), subcostal, and apical views (apical four and five chambers) to assess the anatomy and integrity of atrioventricular valves.
Doppler echocardiographyPulsed-wave Doppler of the flow of the mitral and tricuspid valves was obtained from apical four and five chamber views with sample volume placed at the tip of the mitral and tricuspid valves. The transmitral and tricuspid inflow velocities were traced, and early diastolic (E) and late diastolic (A) velocities as well as E/A ratio were measured. Continuous Doppler study of the pulmonary and aortic valves was performed to assess the systolic pressure gradient across those valves. Systolic pulmonary artery pressure was calculated by obtaining peak systolic gradient of tricuspid regurgitation and adding the value of right atrial pressure. PAH is defined as a mean pulmonary artery pressure>25mmHg in all age groups.10
Laboratory investigationsFive mL of venous blood samples were drawn from each subject under a completely aseptic condition in plain gel separator vacutainers. Serum samples were obtained after clotting by putting it in the water bath at 37°C for 20min; then, centrifugation of the blood at 3000rpm for 5min was done. The obtained serum was divided into two tubes; one was stored at −70°C for VEGF analyses and the second one was analyzed for serum ferritin.
Serum VEGF assessment was done using Human VEGF ELISA Kits from Elabscience Biotechnology Co., Ltd. The detection limit of the VEGF assay was 9pg/mL; the intra-assay precision was ≤6%; and the inter-assay precision was ≤10%. Serum ferritin was measured with the enzyme-linked immunosorbent assay (ELISA) method (Padian Elm Co). Routine laboratory investigations such as complete blood count and liver and kidney function tests were assessed.
Statistical analysisStatistical analysis was done using the Statistical Package for Social Sciences (version 19.0; SPSS Inc., Chicago, IL, USA). Quantitative data were expressed as mean±SD. Differences between two groups were analyzed using independent Student's t-test and Mann–Whitney U test. Differences between more than two groups were analyzed with the one-way analysis of variance (ANOVA) test, and differences between subgroups were analyzed using the post hoc least significant difference (LSD) test. Non-programmatic data were compared by Chi-squared test. Correlations between groups were performed using Spearman correlation coefficients. Receiver operating characteristic curves (ROC) were used to identify the optimal cutoff points of VEGF serum levels for the presence of PAH. Logistic regression analysis was done to determine the risk factors for PAH in children with β-thalassemia major. p-value <0.05 was significant, and in the LSD test p was significant if <0.01.
ResultsThis study included 116 children with β-thalassemia major (70 males and 46 females) on regular blood transfusion. The age of onset of β-thalassemia disorder ranged between 6 and 14 months of age, and the duration of transfusion ranged between 9.5 and 12.5 years. Forty-six of them were subjected to splenectomy. All of them received iron chelation therapy (78 received deferasirox and 38 received deferiprone). In addition, 58 healthy children served as a control group (34 males, 24 females). Their age ranged between 10 and 13 years. We classified children with β-thalassemia into 58 children with PAH and 58 children with a normal range of pulmonary artery pressure.
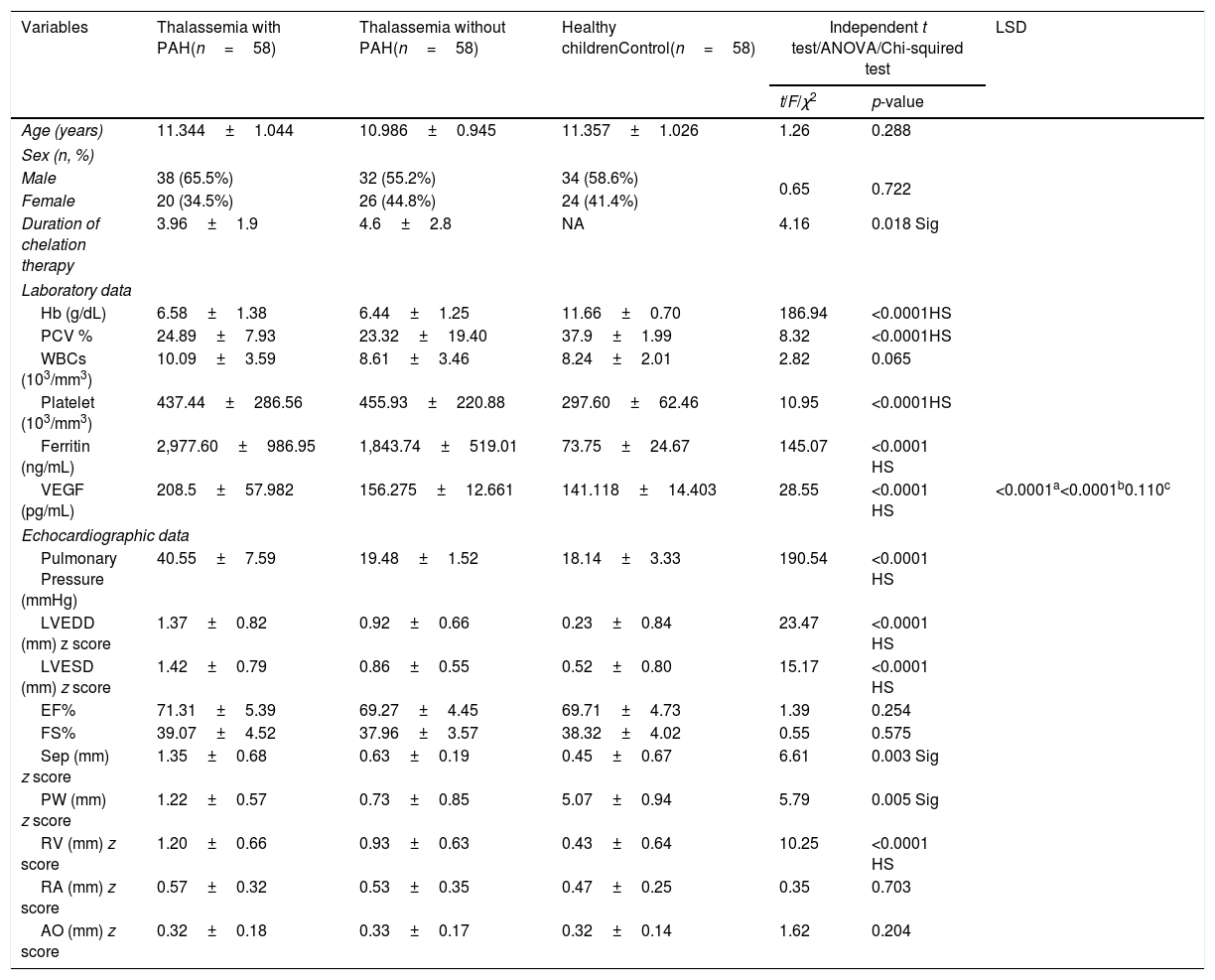
Table 1 shows the demographic, laboratory, and echocardiographic findings of the studied groups. The duration of chelation therapy was significantly lower in thalassemia patients with PAH compared with those without PAH (p=0.002). The hemoglobin and hematocrit values were statistically significantly lower in cases compared to controls (p<0.05). The platelet count and serum ferritin were statistically significantly higher in cases compared to controls (p<0.05). The pulmonary artery pressure, LVEDD, LVESD, PW thickness, septal sickness, and RV diameter were statistically significantly higher in thalassemia patients with PAH compared with those without PAH and control groups (p<0.001). No significant difference was found between all groups of age, sex, WBC count, EF%, FS%, RA diameter and AO diameter (p>0.05). The serum level of VEGF was statistically significantly higher in thalassemia patients with PAH compared with those without PAH and control groups (p<0.001). Further analysis using post hoc LSD illustrated no significant difference in serum levels of VEGF between thalassemia patients without PAH and the control group (p=0.110).
Comparison of clinical, laboratory and echocardiographic data of the studied children.
| Variables | Thalassemia with PAH(n=58) | Thalassemia without PAH(n=58) | Healthy childrenControl(n=58) | Independent t test/ANOVA/Chi-squired test | LSD | |
|---|---|---|---|---|---|---|
| t/F/χ2 | p-value | |||||
| Age (years) | 11.344±1.044 | 10.986±0.945 | 11.357±1.026 | 1.26 | 0.288 | |
| Sex (n, %) | ||||||
| Male | 38 (65.5%) | 32 (55.2%) | 34 (58.6%) | 0.65 | 0.722 | |
| Female | 20 (34.5%) | 26 (44.8%) | 24 (41.4%) | |||
| Duration of chelation therapy | 3.96±1.9 | 4.6±2.8 | NA | 4.16 | 0.018 Sig | |
| Laboratory data | ||||||
| Hb (g/dL) | 6.58±1.38 | 6.44±1.25 | 11.66±0.70 | 186.94 | <0.0001HS | |
| PCV % | 24.89±7.93 | 23.32±19.40 | 37.9±1.99 | 8.32 | <0.0001HS | |
| WBCs (103/mm3) | 10.09±3.59 | 8.61±3.46 | 8.24±2.01 | 2.82 | 0.065 | |
| Platelet (103/mm3) | 437.44±286.56 | 455.93±220.88 | 297.60±62.46 | 10.95 | <0.0001HS | |
| Ferritin (ng/mL) | 2,977.60±986.95 | 1,843.74±519.01 | 73.75±24.67 | 145.07 | <0.0001 HS | |
| VEGF (pg/mL) | 208.5±57.982 | 156.275±12.661 | 141.118±14.403 | 28.55 | <0.0001 HS | <0.0001a<0.0001b0.110c |
| Echocardiographic data | ||||||
| Pulmonary Pressure (mmHg) | 40.55±7.59 | 19.48±1.52 | 18.14±3.33 | 190.54 | <0.0001 HS | |
| LVEDD (mm) z score | 1.37±0.82 | 0.92±0.66 | 0.23±0.84 | 23.47 | <0.0001 HS | |
| LVESD (mm) z score | 1.42±0.79 | 0.86±0.55 | 0.52±0.80 | 15.17 | <0.0001 HS | |
| EF% | 71.31±5.39 | 69.27±4.45 | 69.71±4.73 | 1.39 | 0.254 | |
| FS% | 39.07±4.52 | 37.96±3.57 | 38.32±4.02 | 0.55 | 0.575 | |
| Sep (mm) z score | 1.35±0.68 | 0.63±0.19 | 0.45±0.67 | 6.61 | 0.003 Sig | |
| PW (mm) z score | 1.22±0.57 | 0.73±0.85 | 5.07±0.94 | 5.79 | 0.005 Sig | |
| RV (mm) z score | 1.20±0.66 | 0.93±0.63 | 0.43±0.64 | 10.25 | <0.0001 HS | |
| RA (mm) z score | 0.57±0.32 | 0.53±0.35 | 0.47±0.25 | 0.35 | 0.703 | |
| AO (mm) z score | 0.32±0.18 | 0.33±0.17 | 0.32±0.14 | 1.62 | 0.204 | |
Sig, significant; HS, highly significant; LSD, least significant difference; PAH, pulmonary arterial hypertension; Hb, hemoglobin; PCV, packed cell volume; WBCs, white blood cells; VEGF, vascular endothelial growth factor; LVEED, left ventricle end diastolic dimension; LVESD, left ventricle end systolic dimension; EF%, ejection fraction; FS%, fractional shortening; Sep, septum; PW, posterior wall; RA, right atrium; RV, right ventricle; AO, aorta.
The serum level of VEGF at a cutoff point of >169pg/mL had a sensitivity of 93.1% and a specificity of 93.1% for the presence of PAH in children with β-thalassemia major, as demonstrated in the ROC curve study (Fig. 1).

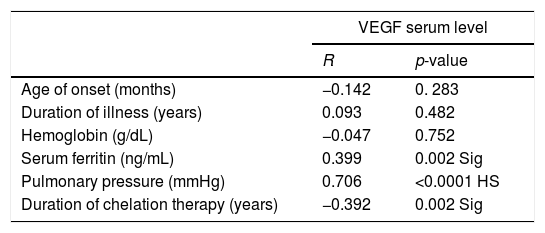
Spearman correlation analysis showed that VEGF serum level had a significant positive correlation with serum ferritin and pulmonary artery pressure (r=0.399, p=0.002; r=0.706, p<0.001, respectively). Additionally, there was a significant negative correlation between VEGF serum level and the duration of iron chelation therapy in children with β-thalassemia major as demonstrated (r=0.392, p=0.002) in Table 2.
Correlation between VEGF serum level and other parameters.
| VEGF serum level | ||
|---|---|---|
| R | p-value | |
| Age of onset (months) | −0.142 | 0. 283 |
| Duration of illness (years) | 0.093 | 0.482 |
| Hemoglobin (g/dL) | −0.047 | 0.752 |
| Serum ferritin (ng/mL) | 0.399 | 0.002 Sig |
| Pulmonary pressure (mmHg) | 0.706 | <0.0001 HS |
| Duration of chelation therapy (years) | −0.392 | 0.002 Sig |
Sig, significant; HS, highly significant; VEGF, vascular endothelial growth factor.
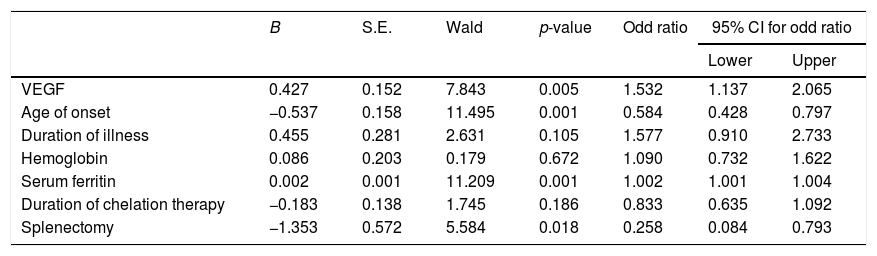
Logistic regression analysis demonstrated that there was a high statistical significant association between the age of onset ≤6 months (OR=0.5; 95% CI, 0.428–0.797; p=0.001), serum ferritin >1000ng/mL (OR=1.002; 95% CI, 1.001–2.004; p=0.001), elevated VEGF ≥169pg/mL (OR=1.5; 95% CI, 1.137–2.065; p=0.005) and splenectomy (OR=0.258; 95% CI, 0.084–0.793; p=0.018) and the development of PAH in children with β-thalassemia major as demonstrated in Table 3.
Logistic regression analysis of risk factors for PAH in children with β-thalassemia major.
| B | S.E. | Wald | p-value | Odd ratio | 95% CI for odd ratio | ||
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| VEGF | 0.427 | 0.152 | 7.843 | 0.005 | 1.532 | 1.137 | 2.065 |
| Age of onset | −0.537 | 0.158 | 11.495 | 0.001 | 0.584 | 0.428 | 0.797 |
| Duration of illness | 0.455 | 0.281 | 2.631 | 0.105 | 1.577 | 0.910 | 2.733 |
| Hemoglobin | 0.086 | 0.203 | 0.179 | 0.672 | 1.090 | 0.732 | 1.622 |
| Serum ferritin | 0.002 | 0.001 | 11.209 | 0.001 | 1.002 | 1.001 | 1.004 |
| Duration of chelation therapy | −0.183 | 0.138 | 1.745 | 0.186 | 0.833 | 0.635 | 1.092 |
| Splenectomy | −1.353 | 0.572 | 5.584 | 0.018 | 0.258 | 0.084 | 0.793 |
VEGF, vascular endothelial growth factor; PAH, pulmonary artery hypertension; B, coefficient; S.E., standard error; Wald, Wald Chi-squared test; CI, confidence interval.
In this study, we found 46 cases of splenectomy and 70 cases without splenectomy. Regarding the effect of splenectomy, PAH and VEGF were statistically significantly higher in post-splenectomy patients [p=0.007, p=0.011; respectively].
DiscussionProper management of children with β-thalassemia improves their quality of life and decreases comorbidities.11 Endothelial activation plays an important role in the pathogenesis of PAH through vascular remodeling and angiogenesis.3 PAH represents a common complication of β-thalassemia, with an incidence ranging from 10% to 75%.12 We hypothesized that an elevated serum level of VEGF may reflect the severity of pulmonary vascular remodeling that is involved in the development of PAH.
Pulmonary hypertension is a progressive multifactorial disorder. Our study demonstrated significant association between younger age of onset, elevated VEGF, serum ferritin levels, splenectomy, and the development of PAH in thalassemia cases.
VEGF is an important regulator of endothelial proliferations, vascular remodeling, and angiogenesis that is involved in the development and progression of PAH.13,14 The role of VEGF over-expression in the development of PAH has been widely studied in animal models and in non-hematological disorders.15,16 Chronic anemia decreases both the oxygen tension and the oxygen-carrying capacity, leading to a steady state of tissue hypoxia which is a potent stimulator of VEGF mRNA expression.17 Additionally, VEGF elevation reflects vascular activation that may be mediated by the toxic effect of chelation therapy or iron overload. Several studies have demonstrated that serum level of VEGF was elevated in patients with chronic hemolytic anemia, including thalassemia either alone or with other angiogenic cytokines and tumor necrosis factor.9,18,19
Our findings highlight a significant association between elevated VEGF and PAH in such patients with high sensitivity (93.1%) and specificity (93.1%) at a cutoff point of >169pg/mL. VEGF serum level has a significant positive correlation with pulmonary pressure. Furthermore, elevated VEGF >169pg/mL was associated with 1.5 times higher risk for development of PAH.
Iron overload contributes to the development of PAH through several mechanisms, including iron deposition in lung parenchyma, which induces lung interstitial fibrosis and increases free radicals and oxidative stress, which in turn trigger many inflammatory cascades causing pulmonary vascular injury.20 Iron overload was evaluated by measuring serum ferritin and showed significant positive correlation with pulmonary pressure, which agrees with other reports in both transfusion-dependent and transfusion-independent β-thalassemia.21,22
Previous studies reported that early and regular use of iron chelation had lowered the incidence of pulmonary hypertension in β-thalassemia by decreasing iron overload.11,23 Animal models revealed that iron chelation decreases lung iron deposition and prevents pulmonary complications in subjects with thalassemia.24 However, this finding disagrees with our results, which showed no significant correlation between duration of chelation therapy and development of PAH. This can be explained by a shorter length of iron chelation therapy and poor compliance in our study in comparison to other studies.
In the current study, PAH was statistically significantly higher in post-splenectomy patients. Similarly to our findings, Chueamuangphan et al.25 have illustrated the strong association between splenectomy and the increased risk of PAH in patients with β-thalassemia. Several mechanisms were suggested to explain this association including thrombocytosis, platelet activation, increased microparticles, nucleated red blood cells, upregulation of vascular adhesion molecules and hyper-coagulable state in post-splenectomy patients, which induce thrombotic obliteration of the pulmonary vasculature.24,26
Furthermore, this study showed that VEGF serum level had a positive correlation with the serum ferritin in children with β-thalassemia (p<0.002). In contrast to our results, other studies reported no significant correlation between serum VEGF and ferritin levels.18,19,27
In the current study, the serum VEGF had a negative correlation with length of iron chelation therapy (r=−0.392, p<0.002). Studies that explored the impact of iron chelation on VEGF expression showed variable results that either agree with our findings18,28 or found no significant correlation.11,19 This could be explained by a different length of iron chelation therapy affecting iron overload.
Our study demonstrated a significant elevation in serum level of VEGF in post-splenectomy thalassemia cases that was higher than in non-splenectomy thalassemia cases, which coincides with Abdel-Aziz et al.9 Olgar et al.27 reported that splenectomy and longer duration of chelation therapy are negatively correlated with VEGF serum level, while younger age of initiation of transfusion and frequent blood transfusion had a positive correlation with VEGF serum level in thalassemia patients. However, the previous study revealed no significant correlations between hemoglobin levels and VEGF serum levels, thus agreeing with our findings. Post-splenectomy platelet activation may contribute to increased VEGF levels in peripheral blood.28
The VEGF serum level was higher in β-thalassemia major patients with PAH than in those without PAH and in control groups. The VEGF had a positive correlation with serum ferritin and pulmonary artery pressure. The serum level of VEGF at a cutoff point of >169pg/mL had a sensitivity of 93.1% and specificity of 93.1% for the presence of PAH in children with β-thalassemia major. We recommend that further studies on the same subject, on a large scale of patients, evaluate pulmonary hypertension by catheterization which is more accurate than echocardiography, and encourage good care, proper management and follow-up of our β-thalassemia patients without PAH to prevent them from developing this potentially fatal complication.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Alkholy UM, Mohamed SA, Elhady M, Attar SE, Abdalmonem N, Zaki A. Vascular endothelial growth factor and pulmonary hypertension in children with beta thalassemia major. J Pediatr (Rio J). 2019;95:593–9.




