

The purpose of this study was to illustrate the association between vascular endothelial growth factor level and pulmonary artery hypertension in children with β‐thalassemia major.
MethodThis case–control study was conducted on 116 children with β‐thalassemia major; 58 of them had pulmonary artery hypertension. They were compared to 58 healthy children who were age and sex‐matched (control group). Serum levels of vascular endothelial growth factor and echocardiographic assessment were done for all children.
ResultsVascular endothelial growth factor serum level was significantly higher in children with β‐thalassemia major with pulmonary artery hypertension than in those without pulmonary artery hypertension, as well as in control groups (p<0.001). Vascular endothelial growth factor serum level had a significant positive correlation with pulmonary artery pressure and serum ferritin, as well as a significant negative correlation with the duration of chelation therapy. Logistic regression analysis revealed that elevated vascular endothelial growth factor (Odd Ratio=1.5; 95% Confidence Interval, 1.137–2.065; p=0.005) was an independent risk factor of pulmonary artery hypertension in such children. Vascular endothelial growth factor serum level at a cutoff point of >169pg/mL had 93.1% sensitivity and 93.1% specificity for the presence of pulmonary artery hypertension in children with β‐thalassemia major.
ConclusionElevated vascular endothelial growth factor serum level is associated with pulmonary artery hypertension in children with β‐thalassemia.
A finalidade deste estudo foi exemplificar a associação entre o nível de fator de crescimento endotelial vascular e a hipertensão arterial pulmonar em crianças com talassemia beta maior.
MétodoEste estudo caso‐controle foi realizado em 116 crianças com talassemia beta maior; 58 das quais apresentaram hipertensão arterial pulmonar em comparação com 58 crianças saudáveis pareadas por idade e sexo (grupo de controle). Os níveis séricos do fator de crescimento endotelial vascular e a avaliação ecocardiográfica foram realizados em todas as crianças.
ResultadosO nível sérico do fator de crescimento endotelial vascular foi significativamente maior em crianças com talassemia beta maior com hipertensão arterial pulmonar que as crianças sem hipertensão arterial pulmonar e os grupos de controle (p<0,001). O nível sérico do fator de crescimento endotelial vascular apresentou uma correlação positiva significativa com a pressão arterial pulmonar e a ferritina sérica e correlação negativa significativa com a duração da terapia de quelação. A análise de regressão logística revelou que o fator de crescimento endotelial vascular elevado (RC=1,5; IC de 95%: 1,137‐2,065; p=0,005) foi um fator de risco independente de hipertensão arterial pulmonar nessas crianças. O nível sérico do fator de crescimento endotelial vascular no ponto de corte> 169 (pg/mL) apresentou 93,1% de sensibilidade e 93,1% de especificidade na presença de hipertensão arterial pulmonar em crianças com talassemia beta maior.
ConclusãoO nível sérico do fator de crescimento endotelial vascular elevado está associado à hipertensão arterial pulmonar em crianças com talassemia beta.
A disfunção miocárdica e a insuficiência cardíaca foram consideradas causas comuns de morbidez e mortalidade em crianças com talassemia beta maior, que foram relacionadas principalmente a hemólise crônica e sobrecarga de ferro.1 Contudo, a hipertensão arterial pulmonar (HAP) surgiu como um importante fator de risco de função ventricular direita prejudicada desses pacientes.1 A HAP é uma doença vascular multifatorial e complexa. O principal mecanismo subjacente da HAP em pacientes talassemia beta ainda não é entendido. Foram sugeridos vários fatores que interagem, inclusive ativação plaquetária, hemólise crônica, angiogênese disfuncional, estresse oxidativo e sobrecarga de ferro.2 O processo de hemólise crônica na talassemia causa mais produção de hemoglobina livre, que elimina o óxido nítrico que protege contra o dano endotelial vascular e libera enzima arginase na circulação, converte L‐arginina para ornitina, passa, assim, a produzir óxido nítrico, que também serve de mediador para relaxamento vascular.3 A HAP é uma das doenças proangiogênicas; contudo, o papel dos fatores angiogênicos, como o fator de crescimento endotelial vascular (VEGF) na patogênese de HAP, não é bem demonstrado.
A ecocardiografia convencional é uma ferramenta confiável, não invasiva, para detecção e acompanhamento da pressão arterial pulmonar; contudo, a cateterização cardíaca é o método padrão de base de diagnóstico de HAP.4 Além desses métodos de diagnóstico tradicionais, os biomarcadores séricos relacionados à cepa miocárdica ou à patologia subjacente têm emergido amplamente.5 Como a HAP é uma doença progressiva, irreversível com risco de vida, há urgência de uma ferramenta fácil, disponível e subjetiva para avaliar o risco e a progressão individualizados da HAP em crianças com talassemia beta maior.
As células endoteliais vasculares pulmonares são um importante regulador de tônus vascular. O VEGF é um peptídeo angiogênico potente que regula a proliferação do endotélico vascular e a permeabilidade vascular e promove neovascularização.6 Suspeitou‐se que o nível elevado no plasma do VEGF é um dos mecanismos envolvidos na remodelação vascular e no desenvolvimento de HAP.7 A upregulation (suprarregulação) do VEGF foi induzida ao expor pacientes com anemia hemolítica crônica a hipóxia.8 Estudos anteriores relataram elevação das citocinas angiogênicas na talassemia, como VEGF, fator de crescimento de fibroblasto básico, angiogenina, angiopoetina ou VEGF e fator de necrose tumoral.9
Na tentativa de melhorar nosso entendimento do mecanismo patofisiológico e dos fatores de risco de HAP em crianças com talassemia beta maior, fizemos este estudo para exemplificar a associação com o nível sérico do VEGF e a HAP nessas crianças. Além disso, para explorar a relação entre os níveis séricos do VEGF com os dados clínicos, as características de quelação e a ferritina sérica nas crianças com talassemia beta maior.
MétodosEste estudo caso‐controle comparativo foi feito em 116 crianças com diagnóstico confirmado de talassemia beta maior que receberam transfusão sanguínea regular de setembro de 2015 a janeiro de 2017; 58 delas apresentaram hipertensão pulmonar (média da pressão arterial pulmonar> 25mmHg) confirmada por ecocardiografia, ao passo que o restante das 58 crianças apresentou pressão arterial pulmonar normal. As crianças que atenderam a nossos critérios de inclusão foram selecionadas consecutivamente do Departamento de Pediatria do Hospital Universitário Al‐Zahraa, Al‐Azhar University, Cairo, Egito. Além disso, 58 crianças saudáveis (grupo de controle) que não apresentaram doença hematológica confirmada por hemograma completo, eletroforese de hemoglobina e não apresentaram anomalias cardíacas confirmadas por ecocardiografia e que atenderam aos mesmos critérios de exclusão do grupo de pacientes. Elas foram selecionadas a partir da idade e sexo pareados das crianças com talassemia. O consentimento informado por escrito foi obtido dos responsáveis pelas crianças e a aprovação foi fornecida pelo Comitê de Ética da Faculdade de Medicina.
Os critérios de inclusão foram crianças com talassemia beta maior confirmada por eletroforese de hemoglobina e suas idades variaram de 10 a 13 anos. Elas receberam uma transfusão sanguínea regular a cada 1‐2 meses na quantidade média de 150‐250 cc de células vermelhas do sangue (CVSs) em cada transfusão. As crianças com doença cardíaca congênita reumática, insuficiência cardíaca, doenças crônicas sistêmicas, exceto talassemia, ou qualquer doença aguda concomitante foram excluídas do estudo.
Histórico e exame clínicoFoi coletado o histórico detalhado e feito exame clínico abrangente em todas as crianças. Os dados em detalhes foram registrados com relação aos dados demográficos, a idade no início da doença, a frequência de transfusão sanguínea, a idade no início e o tipo de terapia de quelação de ferro, o histórico de esplenectomia e quaisquer sintomas de congestão pulmonar, débito cardíaco baixo ou insuficiência cardíaca. Os achados dos exames sistêmico, abdominal e cardíaco foram registrados em detalhes.
Os níveis séricos do VEGF e a avaliação ecocardiográfica foram feitos em todas as crianças incluídas. Avaliamos a associação entre o nível sérico do VEGF e da HAP em crianças com talassemia beta maior e sua relação com outros dados clínicos e laboratoriais.
EcocardiografiaTodos os pacientes e crianças do grupo de controle foram encaminhados para ecocardiografia (Sistema Philips, modelo HD7.XE, Amsterdã). A avaliação foi feita com vários transdutores que variam de 3,5 a 7 MHZ com registro eletrocardiográfico simultâneo para possibilitar tempo de fluxo. Foi usada a ecocardiografia modo M e 2D na posição lateral supina e esquerda para estimar a pressão pulmonar e determinar o diâmetro das câmaras cardíacas.
Medidas do modo MO modo M foi feito do eixo curto paraesternal do ventrículo esquerdo ao aplicar a linha do cursor perpendicular no nível médio papilar para estimar o que segue: dimensões internas do ventrículo esquerdo no fim da sístole e diástole (LVESD, LVEDD) respectivamente, fração de encurtamento (% FS), fração de ejeção (% EF), diâmetro do ventrículo direito (VD), espessamento da parede posterior (PP), espessamento da parede septal, diâmetro da aorta (DA) e diâmetro do átrio direito (AD). O valor normal do % EF varia de faixas etárias de 55‐65% nas crianças.
Eco bidimensional (eco 2D): as imagens de eco 2D foram obtidas das vistas paraesternal (eixo longo e curto), subcostal e apical (quatro apical e cinco câmaras) para avaliar a anatomia e a integridade das válvulas atrioventriculares.
Ecocardiografia DopplerO Doppler de onda pulsada do fluxo das válvulas mitral e tricúspide foi obtido das vistas apicais de quatro e cinco câmaras com amostra de volume posicionada na ponta das valvas mitral e tricúspide, a velocidade de influxo transmital e tricúspide foi traçada e foram medidas as velocidades disastólica precoce (E), diastólica tardia (A) e a razão E/A. O estudo do Doppler contínuo das válvulas pulmonares e aórticas foi feito para avaliar o gradiente pressórico sistólico em todas as válvulas pulmonares e aórticas. A pressão arterial pulmonar sistólica foi calculada ao obter o gradiente sistólico máximo da regurgitação tricúspide e acrescentando o valor da pressão atrial direita. A HAP é definida como a média da pressão arterial pulmonar> 25mm Hg em todas as faixas etárias.10
Investigações laboratoriaisForam coletados 5mL de amostras de sangue venoso de cada indivíduo, em uma condição asséptica completa, em tubos Vacutainers com gel separador. As amostras de soro foram obtidas após coagulação ao colocá‐las em água a 37°C por 20 minutos e serem então centrifugadas a 3.000 rpm por cinco minutos. O soro obtido foi dividido em dois tubos, um foi armazenado a ‐70°C para análises do VEGF e o segundo foi analisado para ferritina no soro.
A avaliação de VEGF no soro foi feita com os Kits Elisa VEGF para Humanos (Elabscience Biotechnology Co, Ltd, EUA). O limite de detecção do ensaio VEGF foi 9 pg/mL, a precisão intraensaio foi ≤ 6 e a precisão interensaio foi ≤ 10%. A ferritina sérica foi medida com o método de ensaio imunossorvente ligado à enzima (Elisa) (Padian Elm Co, Ltd, EUA). Foram avaliadas as investigações laboratoriais de rotina, como hemograma completo, testes da função hepática e renal.
Análise estatísticaA análise estatística foi feita com a versão 19.0 do Pacote Estatístico para as Ciências Sociais (SPSS Inc., Chicago, IL, EUA). Os dados quantitativos foram expressos como média±DP. As diferenças entre os dois grupos foram analisadas com o teste t de Student e o teste U de Mann Whitney independentes. A diferença entre mais de dois grupos foi avaliada com a análise de variância unidirecional (Anova) e as diferenças entre os subgrupos foram analisadas pelo teste de diferença mínima significativa post hoc (DMS). Os dados não programáticos foram comparados com o teste qui‐quadrado. As correlações entre os grupos foram feitas com os coeficientes de correlação de Spearman. As curvas da característica de operação do receptor (ROC) foram usadas para identificar os pontos de corte ideais dos níveis séricos do VEGF na presença de HAP. Foi feita a análise de regressão logística para determinar os fatores de risco para HAP em crianças com talassemia beta maior. O valor de p <0,05 foi significativo e no teste de DMS foi significativo se <0,01.
ResultadosEste estudo incluiu 116 crianças com talassemia beta maior (70 meninos e 46 meninas) em transfusão sanguínea regular. A idade de início da talassemia beta variou entre 6‐14 meses e a duração da transfusão variou entre 9,5‐12,5 anos; 46 delas foram submetidas a esplenectomia. Todas elas receberam terapia de quelação de ferro (78 receberam deferasirox e 38 receberam deferiprona). Além disso, 58 crianças saudáveis atuaram como grupo de controle (34 meninos, 24 meninas). Sua idade variou entre 10‐13 anos. Classificamos as crianças com talassemia beta em 58 crianças com HAP e 58 crianças com valor normal de pressão arterial pulmonar.
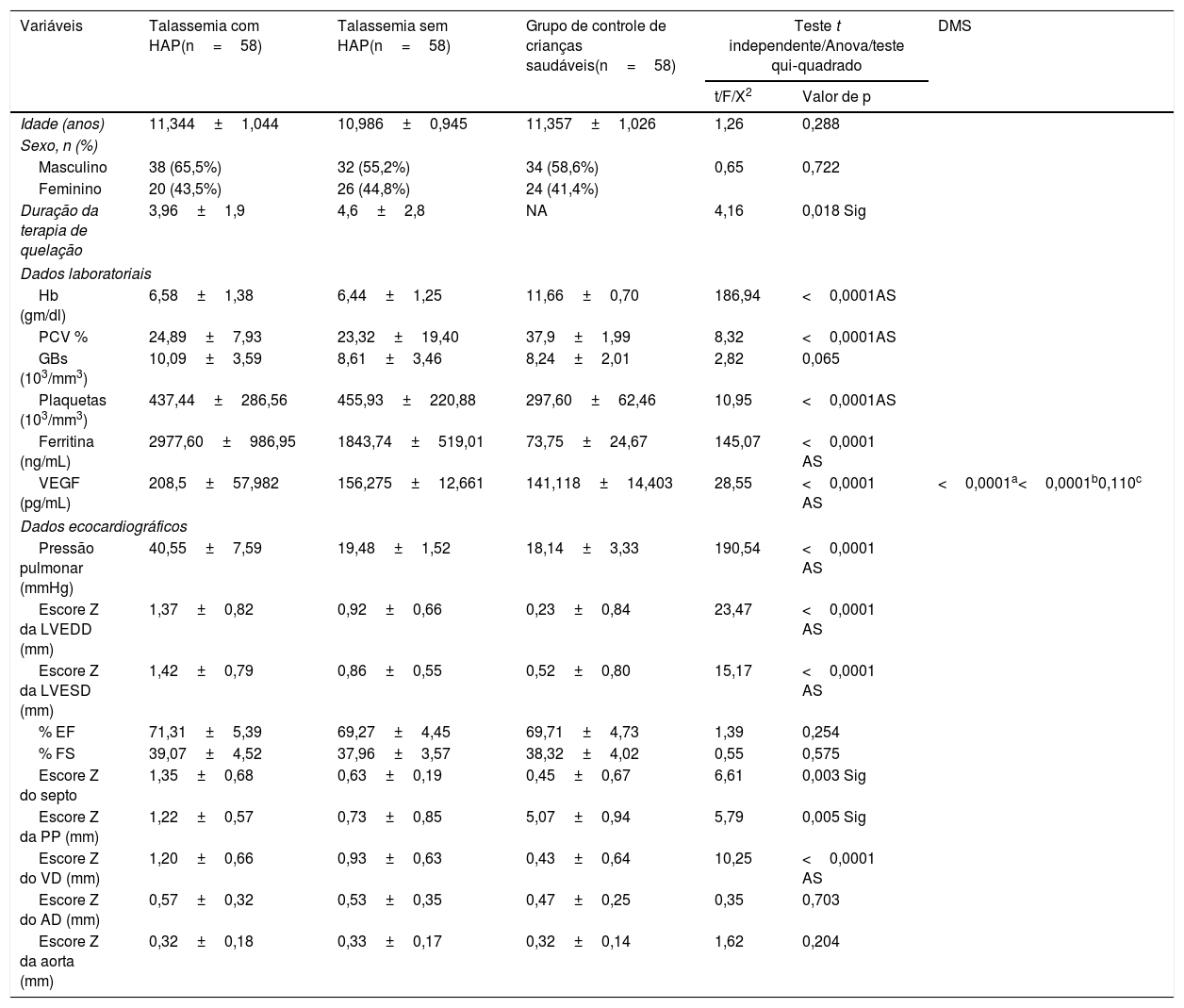
A tabela 1 mostra os achados demográficos, laboratoriais e ecocardiográficos dos grupos do estudo. A duração da terapia de quelação foi significativamente menor em pacientes com talassemia com HAP em comparação com os sem HAP (p=0,002). A hemoglobina e o valor de hematócritos foram estatística e significativamente menores nos casos comparados com os grupos de controle (p <0,05). A contagem de plaquetas e a ferritina sérica foram estatística e significativamente maiores nos casos comparados com os grupos de controle (p <0,05). A pressão arterial pulmonar, LVEDD, LVESD, espessura da PP, doença do septo e diâmetro do VD foram estatística e significativamente maiores em pacientes com talassemia com HAP em comparação com os sem HAP e aos grupos de controle (p <0,001). Não foi encontrada diferença significativa entre todas as faixas etárias, sexo, contagem de leucócitos, % EF, % FS, diâmetro do AD e diâmetro da aorta (p> 0,05). O nível sérico do VEGF foi significativamente maior em pacientes com talassemia com HAP em comparação com os sem HAP e os grupos de controle (p <0,001). Uma análise adicional com a DMS post hoc não mostrou diferença significativa nos níveis séricos do VEGF entre os pacientes com talassemia sem HAP e o grupo de controle (p=0,110).
Comparação dos dados clínicos, laboratoriais e ecocardiográficos das crianças incluídas no estudo
| Variáveis | Talassemia com HAP(n=58) | Talassemia sem HAP(n=58) | Grupo de controle de crianças saudáveis(n=58) | Teste t independente/Anova/teste qui‐quadrado | DMS | |
|---|---|---|---|---|---|---|
| t/F/X2 | Valor de p | |||||
| Idade (anos) | 11,344±1,044 | 10,986±0,945 | 11,357±1,026 | 1,26 | 0,288 | |
| Sexo, n (%) | ||||||
| Masculino | 38 (65,5%) | 32 (55,2%) | 34 (58,6%) | 0,65 | 0,722 | |
| Feminino | 20 (43,5%) | 26 (44,8%) | 24 (41,4%) | |||
| Duração da terapia de quelação | 3,96±1,9 | 4,6±2,8 | NA | 4,16 | 0,018 Sig | |
| Dados laboratoriais | ||||||
| Hb (gm/dl) | 6,58±1,38 | 6,44±1,25 | 11,66±0,70 | 186,94 | <0,0001AS | |
| PCV % | 24,89±7,93 | 23,32±19,40 | 37,9±1,99 | 8,32 | <0,0001AS | |
| GBs (103/mm3) | 10,09±3,59 | 8,61±3,46 | 8,24±2,01 | 2,82 | 0,065 | |
| Plaquetas (103/mm3) | 437,44±286,56 | 455,93±220,88 | 297,60±62,46 | 10,95 | <0,0001AS | |
| Ferritina (ng/mL) | 2977,60±986,95 | 1843,74±519,01 | 73,75±24,67 | 145,07 | <0,0001 AS | |
| VEGF (pg/mL) | 208,5±57,982 | 156,275±12,661 | 141,118±14,403 | 28,55 | <0,0001 AS | <0,0001a<0,0001b0,110c |
| Dados ecocardiográficos | ||||||
| Pressão pulmonar (mmHg) | 40,55±7,59 | 19,48±1,52 | 18,14±3,33 | 190,54 | <0,0001 AS | |
| Escore Z da LVEDD (mm) | 1,37±0,82 | 0,92±0,66 | 0,23±0,84 | 23,47 | <0,0001 AS | |
| Escore Z da LVESD (mm) | 1,42±0,79 | 0,86±0,55 | 0,52±0,80 | 15,17 | <0,0001 AS | |
| % EF | 71,31±5,39 | 69,27±4,45 | 69,71±4,73 | 1,39 | 0,254 | |
| % FS | 39,07±4,52 | 37,96±3,57 | 38,32±4,02 | 0,55 | 0,575 | |
| Escore Z do septo | 1,35±0,68 | 0,63±0,19 | 0,45±0,67 | 6,61 | 0,003 Sig | |
| Escore Z da PP (mm) | 1,22±0,57 | 0,73±0,85 | 5,07±0,94 | 5,79 | 0,005 Sig | |
| Escore Z do VD (mm) | 1,20±0,66 | 0,93±0,63 | 0,43±0,64 | 10,25 | <0,0001 AS | |
| Escore Z do AD (mm) | 0,57±0,32 | 0,53±0,35 | 0,47±0,25 | 0,35 | 0,703 | |
| Escore Z da aorta (mm) | 0,32±0,18 | 0,33±0,17 | 0,32±0,14 | 1,62 | 0,204 | |
% EF, fração de ejeção; % FS, fração de encurtamento; AD, átrio direito; AS, altamente significativo; DMS, diferença menos significativa; GBs, glóbulos brancos; HAP, hipertensão arterial pulmonar; Hb, hemoglobina; LVEED, dimensão diastólica final do ventrículo esquerdo; LVESD, dimensão sistólica final do ventrículo esquerdo; PCV, volume de células compactadas; PP, parede posterior; Sig, significativo; VD, ventrículo direito; VEGF, fator de crescimento endotelial vascular.
O nível sérico do VEGF no ponto de corte> 169 (pg/mL) apresentou sensibilidade de 93,1% e especificidade de 93,1% na presença de HAP em crianças com talassemia beta maior, conforme demonstrado no estudo da curva ROC, figura 1.
 com área abaixo da curva de 0,956 apresentou sensibilidade de 93,1% e especificidade de 93,1% na presença de HAP em crianças com talassemia beta maior.")
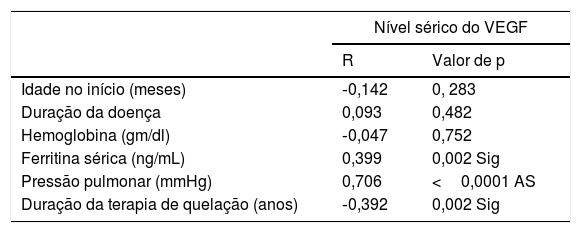
A análise da correlação de Spearman mostrou que o nível sérico do VEGF apresentou correlação positiva significativa com a ferritina sérica e a pressão arterial pulmonar (r=0,399, p=0,002, r=0,706, p <0,001; respectivamente). Além disso, houve uma correlação negativa significativa entre o nível sérico do VEGF e a duração da terapia de quelação de ferro nas crianças com talassemia beta maior, conforme demonstrado (r=0,392, p=0,002) na tabela 2.
Correlação entre o nível sérico do VEGF e outros parâmetros
| Nível sérico do VEGF | ||
|---|---|---|
| R | Valor de p | |
| Idade no início (meses) | ‐0,142 | 0, 283 |
| Duração da doença | 0,093 | 0,482 |
| Hemoglobina (gm/dl) | ‐0,047 | 0,752 |
| Ferritina sérica (ng/mL) | 0,399 | 0,002 Sig |
| Pressão pulmonar (mmHg) | 0,706 | <0,0001 AS |
| Duração da terapia de quelação (anos) | ‐0,392 | 0,002 Sig |
AS, altamente significativo; Sig, significativo; VEGF, fator de crescimento endotelial vascular.
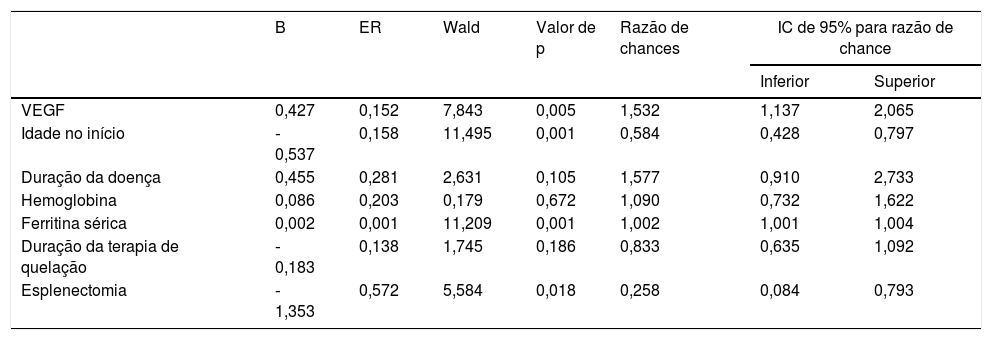
A análise de regressão logística demonstrou que houve uma associação estatística altamente significativa com a idade de início <6 meses (RC=0,5; IC de 95%: 0,428‐0,797; p=0,001), ferritina sérica> 1.000 ng/mL (RC=1,002; IC de 95%: 1,001‐2,004; p=0,001), VEGF elevado ≥ 169 pg/mL (RC=1,5; IC de 95%: 1,137‐2,065; p=0,005) e esplenectomia (RC=0,58; IC de 95%: 0,084‐0,793; p=0,018) com o desenvolvimento de HAP nas crianças com talassemia beta maior, conforme demonstrado na tabela 3.
Análise de regressão logística dos fatores de risco para HAP em crianças com talassemia beta maior
| B | ER | Wald | Valor de p | Razão de chances | IC de 95% para razão de chance | ||
|---|---|---|---|---|---|---|---|
| Inferior | Superior | ||||||
| VEGF | 0,427 | 0,152 | 7,843 | 0,005 | 1,532 | 1,137 | 2,065 |
| Idade no início | ‐0,537 | 0,158 | 11,495 | 0,001 | 0,584 | 0,428 | 0,797 |
| Duração da doença | 0,455 | 0,281 | 2,631 | 0,105 | 1,577 | 0,910 | 2,733 |
| Hemoglobina | 0,086 | 0,203 | 0,179 | 0,672 | 1,090 | 0,732 | 1,622 |
| Ferritina sérica | 0,002 | 0,001 | 11,209 | 0,001 | 1,002 | 1,001 | 1,004 |
| Duração da terapia de quelação | ‐0,183 | 0,138 | 1,745 | 0,186 | 0,833 | 0,635 | 1,092 |
| Esplenectomia | ‐1,353 | 0,572 | 5,584 | 0,018 | 0,258 | 0,084 | 0,793 |
B, coeficiente; ER, erro‐padrão; HAP, hipertensão arterial pulmonar; IC, intervalo de confiança; VEGF, fator de crescimento endotelial vascular; Wald, teste qui‐quadrado de Wald.
Neste estudo, descobrimos que 46 casos foram submetidos a esplenectomia e 70 casos não. Com relação ao efeito da esplenectomia, a HAP e o VEGF foram estatística e significativamente maiores nos pacientes submetidos a esplenectomia [(p=0,007), (p=0,11); respectivamente].
DiscussãoO manejo adequado das crianças com talassemia beta melhora sua qualidade de vida e reduz as comorbidades.11 A ativação endotelial desempenha um papel importante na patogênese da HAP por meio da remodelação vascular e angiogênese.3 A HAP representa uma complicação comum da talassemia beta e a incidência variou de 10% a 75%.12 Trabalhamos com a hipótese de que o nível sérico elevado do VEGF pode refletir a gravidade da remodelação vascular pulmonar envolvida no desenvolvimento de HAP.
A hipertensão pulmonar é uma doença multifatorial progressiva. Nosso estudo demonstrou associação significativa entre a idade de início mais cedo, VEGF elevado, níveis de ferritina sérica, esplenectomia e desenvolvimento de HAP em casos de talassemia.
O VEGF é um importante regulador de proliferações endoteliais, remodelação vascular e angiogênese envolvidas no desenvolvimento e progressão da HAP.13,14 O papel da superexpressão do VEGF no desenvolvimento de HAP foi amplamente estudado em modelos com animais e em doenças não hematológicas.15,16 A anemia crônica reduz a tensão do oxigênio e a capacidade de transporte do oxigênio, leva a um estado estável de hipóxia tecidual, um potente estimulador da expressão do mRNA do VEGF.17 Além disso, o aumento do VEGF reflete a ativação vascular que pode ser mediada pelo efeito tóxico da terapia de quelação ou sobrecarga de ferro. Vários estudos demonstraram que o nível sérico do VEGF foi elevado em pacientes com anemia hemolítica crônica, inclusive talassemia sozinha ou com outras citocinas angiogênicas e fator de necrose tumoral.9,18,19
Nosso achado destaca a associação significativa entre VEGF elevado e HAP nesses pacientes com alta sensibilidade (93,1%) e especificidade (93,1%) no ponto de corte de> 169 (pg/mL). O nível sérico do VEGF tem correlação positiva significativa com a pressão pulmonar. Adicionalmente, VEGF elevado> 169 pg/mL foi associado a 1,5 vez risco mais elevado de desenvolver HAP.
A sobrecarga de ferro contribui para o desenvolvimento de HAP por meio de vários mecanismos, inclusive deposição de ferro no parênquima do pulmão, que induz fibrose intersticial, aumento de radicais livres e estresse oxidativo, que geram muitas cascatas inflamatórias e causam dano vascular.20 A sobrecarga de ferro foi avaliada ao se medir a ferritina sérica, que mostrou correlação positiva significativa com a pressão pulmonar, o que está de acordo com outros relatos sobre talassemia beta dependente de transfusão e independente de transfusão.21,22
Estudos anteriores relataram que o uso precoce e regular de quelação de ferro reduziu a incidência de hipertensão pulmonar na talassemia beta ao reduzir a sobrecarga de ferro.11,23 Os modelos em animais revelaram que a quelação de ferro reduz a deposição de ferro nos pulmões e previne complicações pulmonares em indivíduos com talassemia.24 Porém, isso não está em linha com nossos resultados, que não mostraram correlação significativa entre a duração da terapia de quelação e o desenvolvimento de HAP. Isso pode ser explicado pela duração mais curta do recebimento da terapia de quelação de ferro e pela baixa conformidade em nosso estudo em comparação a outros estudos.
No estudo atual, a HAP foi estatística e significativamente maior nos pacientes submetidos a esplenectomia. Semelhante a nossos achados, Chueamuangphan et al.25 exemplificaram a forte associação entre a esplenectomia e o aumento do risco de HAP em pacientes com talassemia beta. Vários mecanismos foram sugeridos para explicar essa associação, inclusive trombocitose, ativação plaquetária, aumento de micropartículas, glóbulos vermelhos nucleados, upregulation das moléculas de adesão vascular e estado de hipercoagulabilidade em pacientes submetidos a esplenectomia, que induz obliteração trombótica da vasculatura pulmonar.24,26
Além disso, este estudo mostrou que o nível sérico do VEGF apresentou correlação positiva com a ferritina sérica em crianças com talassemia beta (p <0,002). Em contraste com nossos resultados, outros estudos não relataram correlação significativa entre os níveis séricos do VEGF e da ferritina.18,19,27
No estudo atual, o VEGF no soro apresentou uma correlação negativa com a duração do recebimento da terapia de quelação de ferro (r=‐0,392, p <0,002). Estudos que exploraram o impacto da quelação de ferro sobre a expressão do VEGF mostraram resultados variáveis que corroboram nossos achados18,28 ou não encontraram correlação significativa.11,19 Isso pode ser explicado pela diferente duração do recebimento da terapia de quelação de ferro, que afeta a sobrecarga de ferro.
Nosso estudo demonstrou um aumento mais significativo no nível sérico do VEGF nos casos de talassemia pós‐esplenectomia do que nos casos de talassemia não submetidos a esplenectomia, o que coincide com o relatado por Abdel‐Aziz et al.9 e Olgar et al.,27 que a esplenectomia e mais duração da terapia de quelação têm correlação negativa com o nível sérico do VEGF, ao passo que o início precoce da transfusão e a transfusão sanguínea frequente apresentaram correlação positiva com o nível sérico do VEGF em pacientes com talassemia. Contudo, o estudo anterior não revelou correlação significativa entre os níveis de hemoglobina e os níveis séricos do VEGF, o que é compatível com nossos achados. A ativação plaquetária pós‐esplenectomia pode contribuir para o aumento dos níveis do VEGF no sangue periférico.28
O nível sérico do VEGF foi maior em pacientes com talassemia beta maior com HAP que nos pacientes sem HAP e nos grupos de controle. O VEGF apresentou correlação positiva com a ferritina sérica e a pressão arterial pulmonar. O nível sérico do VEGF no ponto de corte> 169 (pg/mL) apresentou sensibilidade de 93,1% e especificidade de 93,1% na presença de HAP em crianças com talassemia beta maior. Recomendamos que haja estudos adicionais sobre o mesmo assunto em grande escala de pacientes, avaliação da hipertensão pulmonar por cateterização, que é mais precisa que a ecocardiografia, e incentivar bom tratamento, manejo adequado e acompanhamento de nossos pacientes com talassemia beta sem HAP para prevenir que eles desenvolvam essa complicação possivelmente fatal.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Alkholy UM, Mohamed SA, Elhady M, Attar SE, Abdalmonem N, Zaki A. Vascular endothelial growth factor and pulmonary hypertension in children with beta thalassemia major. J Pediatr (Rio J). 2019;95:593–9.




