


To review the current evidence base for the diagnosis and management of the childhood epilepsies and to draw attention to the current gaps in this evidence base. The focus will be on therapeutic aspects. Current International League Against Epilepsy (ILAE) terminology will be described and used throughout the discussion. The review will draw attention to recent advances that have been made in both our understanding and treatment of the childhood epilepsies. Potential future directions for research and treatment options will be discussed.
SourcesOriginal articles relevant to the subject were obtained from the MedLine database using pertinent MeSH terms. Relevant papers were read and assimilated. Citation searching was used.
Summary of the findingsEpilepsy is a major cause of global disease burden. Childhood epilepsies are a heterogeneous group of conditions. A multi-axial diagnostic approach should be taken prior to making treatment and management decisions for any individual patient. For the majority of patients, successful control of seizures can be achieved with a single medication. However, a significant minority develops refractory disease. Epilepsy surgery can provide cure for a carefully selected group of these cases.
ConclusionsThere remain significant gaps the evidence base for treatment in several areas of childhood epilepsy. Concerted multi-center efforts should be made to try to close these gaps. A personalized medicine approach may help to reduce the proportion of refractory cases of childhood epilepsy in future.
Analisar a base de evidências atual para o diagnóstico e tratamento das epilepsias da infância e chamar a atenção para as lacunas atuais nessa base de evidências. O foco será os aspectos terapêuticos. A terminologia atual da Liga Internacional contra a Epilepsia (ILAE) será descrita e utilizada na discussão. A análise chamará a atenção para os recentes avanços em nosso entendimento e no tratamento das epilepsias da infância. Serão discutidas possíveis orientações futuras para as opções de pesquisa e tratamento.
Fontes de dadosTrabalhos originais relevantes para o assunto foram obtidos da base de dados MedLine usando termos relevantes do MeSH. Os trabalhos relevantes foram lidos e assimilados. Foi usada pesquisa de citações.
Resumo dos dadosA epilepsia é uma das maiores causas da carga global de doenças. As epilepsias da infância representam um grupo heterogêneo de doenças. Uma abordagem multiaxial do diagnóstico deve ser realizada antes da tomada de decisões de tratamento de qualquer paciente individual. Na maioria dos pacientes, o controle bem-sucedido das crises pode ser obtido com uma única medicação. Contudo, uma minoria significativa desenvolve doença refratária. A cirurgia de epilepsia pode curar um grupo cuidadosamente selecionado desses casos.
ConclusõesAinda existem lacunas significativas na base de evidências de tratamento em diversas áreas de epilepsia da infância. Devem ser envidados esforços multicêntricos concertados para tentar fechar essas lacunas. Uma abordagem médica personalizada pode ajudar a reduzir a proporção de casos refratários de epilepsia da infância no futuro.
Epilepsy is the most prevalent chronic neurological disease in the world, with an estimated 65 million people affected.1 Severe epilepsy ranks as the fourth largest cause of global disease burden.2 Crude incidence estimates for epilepsy vary from 15 to 113 per 100,000 per year, depending on the population studied.3 The incidence of epilepsy in childhood is more than twice that in the adult population.4
DefinitionsEpilepsy has been conceptually defined as a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures. An epileptic seizure is a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.5 To facilitate the practical application of epilepsy as a diagnostic term, the International League Against Epilepsy (ILAE) finalized an operational definition of epilepsy in 2014 (Table 1).6 It is perhaps most helpful to conceptualize epilepsy not as a single condition, but as a group of conditions, “the epilepsies”, reflecting the recognition that the underlying causes of epileptic seizures are both varied and numerous, and that the clinical manifestations of abnormal excessive or synchronous neuronal activity are heterogeneous.
International League Against Epilepsy operational definition of epilepsy 2014.6
| A disease of the brain defined by any of the following conditions |
| 1. At least two unprovoked (or reflex) seizures occurring >24h apart |
| 2. One unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years |
| 3. Diagnosis of an epilepsy syndrome |
When considering a diagnosis of epilepsy, a multi-axial approach is recommended.7 The following axes should be considered:
- 1.
Is the diagnosis epilepsy?
- 2.
What are the seizure types?
- 3.
Can the diagnosis of an established epilepsy syndrome be made?
- 4.
What is the underlying cause for the epilepsy?
- 5.
Are there any comorbidities?
Accuracy of diagnosis is paramount, since a diagnosis of epilepsy can have significant implications for patients, their families, and therapeutic management. However, making a diagnosis of epilepsy can be challenging, since there are a number of paroxysmal conditions for which the clinical history may resemble that of an epileptic seizure, and because there is no definitive diagnostic investigation for this condition. Misdiagnosis rates in epilepsy have traditionally been high. Of 233 children referred to a tertiary epilepsy center in Denmark, 87 (39%) were found by expert assessment not to have epilepsy, and of these 35 (40%) had been started on anti-epileptic drugs (AEDs).8 The most frequently observed differential diagnoses were non-epileptic staring spells (52.8%), psychogenic non-epileptic seizures (10.3%), syncope (3.4%), dystonia (3.4%), and parasomnias (3.4%). Misdiagnosis of non-epileptic events when the true diagnosis is epilepsy appears to be less common. In a Dutch study that included 888 children referred to a tertiary center with paroxysmal events, 19/124 (5.6%) children who had been referred with multiple unclear events were given a diagnosis of epilepsy.9 Initial assessment by a practitioner with expertise and experience in epilepsy diagnosis is thought to significantly reduce misdiagnosis rates.
In many cases, the diagnosis of an epileptic or non-epileptic event can be based on an accurate description of the episode or episodes by the patient and witnesses, with no further investigations necessary. The widespread use of smartphones has greatly facilitated the diagnosis of paroxysmal events. When events are recurrent and there is doubt about their nature, parents should be strongly encouraged to capture them on video. The ILAE has built up an excellent online resource10 where clinicians can view video examples of epileptic and non-epileptic events.
Syncopal events can often be differentiated from epileptic seizures by the patient's description of how they felt before the collapse. Typical prodromal symptoms of syncope are dizziness, visual loss, nausea, sweating, and tinnitus. In all cases or collapse, it is advisable of obtain a 12-lead electrocardiogram, with particular attention to measurement of the corrected QT interval, since cardiac arrhythmia is an important differential.11
Description of seizure typesOnce a clear description of the event(s) has been obtained, if they are thought to be epileptic, the clinician should try to classify the seizure type(s). Table 2 details the current ILAE classification of seizure types. Generalized seizures are conceptualized as beginning within, and rapidly engaging bilateral networks within the brain, whereas focal seizures originate within networks limited to one hemisphere. Focal seizures may progress to engage both hemispheres, resulting in bilateral convulsive features. An electroencephalogram (EEG) is a useful adjunct to seizure type classification, particularly in focal epilepsies for which focal epileptiform activity may help identify a lobar location.
International League Against Epilepsy classification of seizure types.
| Generalized seizures | |
| Generalized tonic–clonic | Bilaterally increased tone, followed by sustained bilateral rhythmic limb jerking. |
| Clonic | Sustained bilateral rhythmic jerking. |
| Absence | |
| Typical absence | Abrupt onset of altered awareness. Memory of events usually impaired. Oral and manual automatisms are common. Clonic movements of parts of the face may occur. |
| Atypical absence | Less abrupt onset and offset than typical absence. May be associated with loss of muscle tone, or subtle myoclonic jerks. Often longer than typical absences. Alteration in awareness variable. |
| Myoclonic absence | Rhythmic myoclonic jerks of the shoulders and arms with tonic abduction, resulting in progressive lifting of the arms during the seizure. The myoclonic jerks are typically bilateral, but may be unilateral or asymmetric. Last 10–60s. Alteration in awareness variable. |
| Absence with eyelid myoclonia | Absence seizures accompanied by brief, repetitive, often rhythmic, fast myoclonic jerks of the eyelids, with simultaneous upward deviation of the eyeballs and extension of the head. Seizures are typically very brief (<6s in duration) and multiple seizures occur on a daily basis. Mostly awareness is retained. |
| Tonic | Bilaterally increased tone of the limbs lasting seconds to a minute. Often occur out of sleep and in runs of varying intensity. The individual is unaware during these events. Individuals may make an expiratory sound at onset. May cause drop attacks. |
| Atonic | Sudden loss or diminution of muscle tone without apparent preceding myoclonic or tonic features. Very brief (<2s) and may involve the head, trunk, or limbs. |
| Myoclonic | |
| Myoclonic | Single or series of jerks (brief muscle contractions). Each jerk is typically milliseconds in duration. |
| Negative myoclonus | Brief cessation of background muscle tone, lasting less than 500ms. The resulting movement produced can have two components, an initial loss of posture caused by the negative myoclonus, and a subsequent voluntary, compensatory movement to restore posture. May occur in isolation or in a series. |
| Myoclonic–atonic | Myoclonic seizure followed by an atonic seizure. Sometimes a series of myoclonic jerks occurs prior to the atonia. The head and limbs are affected, typically resulting in rapid fall. |
| Myoclonic–tonic | Myoclonic seizure followed by a tonic seizure. Sometimes a cluster of myoclonic jerks occurs prior to the increased tone. |
| Focal seizures: can be classified by features, and/or laterality, and/or lobar localization | |
| By features (clinical): one or more type of feature may be present during any single focal seizure | |
| Aura | Auras are subjective and may be sensory or experiential. A sensory aura involves a sensation without an objective clinical sign, which may be visual, auditory, olfactory, epigastric, etc. An experiential aura involves affective, mnemonic, or perceptual subjective phenomena, including depersonalization and hallucinatory events. |
| Motor | A motor feature involves motor activity and may consist of an increase (positive) or decrease (negative) in muscle contraction. |
| Autonomic | Characterized by autonomic phenomena, which can involve cardiovascular, gastrointestinal, vasomotor, and thermoregulatory functions. Examples include palpitations, nausea, butterflies, hunger, chest pain, urge to urinate or defecate, goosebumps, sexual sensation, feeling hot or cold, piloerection, pallor, tachycardia or bradycardia/asystole, flushing, pupillary changes, and lacrimation. |
| Dyscognitive | Involves altered awareness or responsiveness. |
| By hemispheric localization (clinical or electrophysiological) | |
| RightLeft | Indications of hemispheric localization may be subtle, particularly in seizures that rapidly engage bilateral networks. Head turning, eye deviation, and unilateral clonic movements all lateralize to the contralateral hemisphere. |
| By lobar localization (clinical or electrophysiological) | |
| Frontal | Frontal lobe seizures are usually brief. Motor features are prominent. There may be vocalization, bizarre behavior, head and eye deviation, and urinary incontinence. |
| Temporal | Characterized by behavioral arrest and dyscognitive features. Automatisms and auras are common. |
| Parietal | Ictal features are often subjective, and include positive and/or negative sensory features such as paresthesia, disorientation, and complex visual hallucinations. |
| Occipital | Characterized by visual aura, typically multi-colored shapes. |
| Epileptic Spasms | |
| Sudden flexion, extension or mixed flexion-extension of proximal and truncal muscles, lasting 1–2s, i.e., longer than a myoclonic jerk (which lasts milliseconds) but not as long as a tonic seizure (which lasts >2s). Spasms typically occur in a series, usually on wakening. Subtle forms may occur with only chin movement, grimacing, or head nodding. Spasms may be bilaterally symmetric, asymmetric, or unilateral. | |
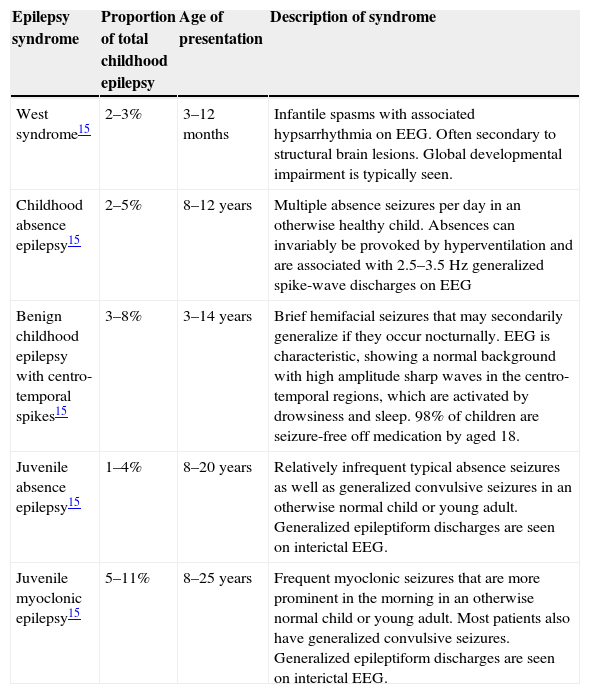
By analyzing the clinical details of a patient's epilepsy, including seizure types, age of onset, and associated comorbidities, it may be possible to diagnose a specific epilepsy syndrome. Many epilepsy syndromes are associated with particular findings on EEG, therefore this investigation can be very helpful in assisting syndrome classification. Achieving a syndromic diagnosis is important, since it may inform therapeutic decisions, further investigations, and prognosis. The ILAE website provides clinical and EEG features of 31 epilepsy syndromes.10 Estimates for the proportion of childhood epilepsy cases that can be diagnosed with an epilepsy syndrome have ranged between 15.7% and 37.1%, depending on the population included.12–14 The most prevalent epilepsy syndromes of childhood appear to be benign epilepsy of childhood with centro-temporal spikes (BCECTS), childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME), and West syndrome (WS). Many of the epilepsies that do not conform to a syndromic diagnosis can still be described in terms of seizure types and etiology (Table 3).
Most frequently encountered childhood epilepsy syndromes.
| Epilepsy syndrome | Proportion of total childhood epilepsy | Age of presentation | Description of syndrome |
|---|---|---|---|
| West syndrome15 | 2–3% | 3–12 months | Infantile spasms with associated hypsarrhythmia on EEG. Often secondary to structural brain lesions. Global developmental impairment is typically seen. |
| Childhood absence epilepsy15 | 2–5% | 8–12 years | Multiple absence seizures per day in an otherwise healthy child. Absences can invariably be provoked by hyperventilation and are associated with 2.5–3.5Hz generalized spike-wave discharges on EEG |
| Benign childhood epilepsy with centro-temporal spikes15 | 3–8% | 3–14 years | Brief hemifacial seizures that may secondarily generalize if they occur nocturnally. EEG is characteristic, showing a normal background with high amplitude sharp waves in the centro-temporal regions, which are activated by drowsiness and sleep. 98% of children are seizure-free off medication by aged 18. |
| Juvenile absence epilepsy15 | 1–4% | 8–20 years | Relatively infrequent typical absence seizures as well as generalized convulsive seizures in an otherwise normal child or young adult. Generalized epileptiform discharges are seen on interictal EEG. |
| Juvenile myoclonic epilepsy15 | 5–11% | 8–25 years | Frequent myoclonic seizures that are more prominent in the morning in an otherwise normal child or young adult. Most patients also have generalized convulsive seizures. Generalized epileptiform discharges are seen on interictal EEG. |
In most epilepsies, investigation for an underlying etiology is warranted, regardless as to whether or not an epilepsy syndrome diagnosis has been made. The exceptions to this rule are typical cases of BCECTS, CAE, JAE, and JME. Although these epilepsies all have a principally genetic etiology, genetic investigation is not currently clinically useful since multiple susceptibility genes are involved and there is no established correlation between genetic findings and either prognosis or therapeutic management. These same syndromes are very rarely associated with structural brain abnormalities, so neuroimaging is also not necessary.15 In all other cases of childhood-onset epilepsy, including atypical cases of the above-listed syndromes, neuroimaging is indicated. The purpose of neuroimaging is to identify any causative structural brain lesion. Some lesions, such as neoplasms, may require immediate management, whilst others, such as focal cortical dysplasia, may prove to be surgically remediable targets further down the line. Abnormal neuroimaging is found in 50% of new childhood-onset epilepsy in which there are focal seizures, and in 15–20% of cases imaging studies provide useful information on etiology and/or seizure focus.16 Magnetic resonance imaging (MRI) is the imaging modality of choice.
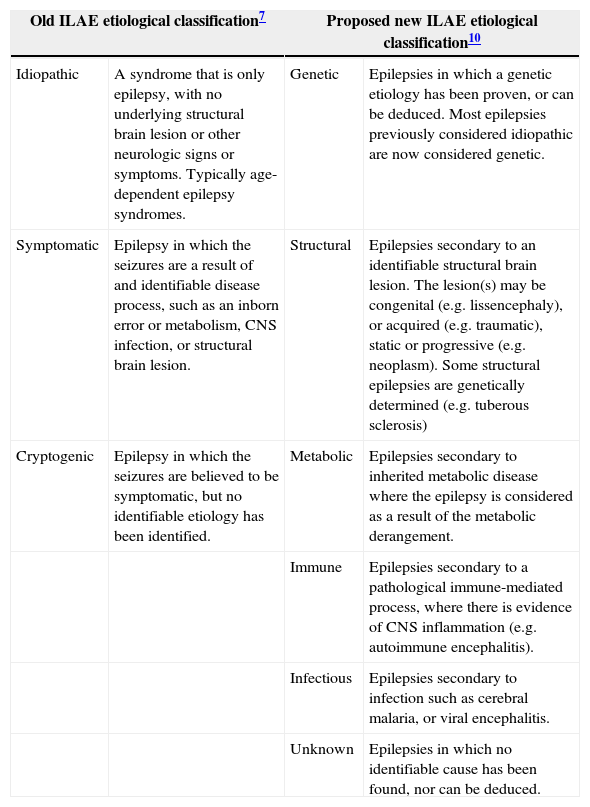
The first significant attempt at reclassifying the epilepsies in line with modern understanding of the disease and its mechanisms was made by the ILAE in 2010. The principal changes to official ILAE nomenclature focused on etiological categorization. The terms “idiopathic,” “symptomatic,” and “cryptogenic” were replaced by the more precise terms “genetic,” “structural,” and “metabolic.” The inclusion of further etiological categories “immune” and “infectious” is currently under discussion (Table 4).
International League Against Epilepsy (ILAE) etiological classification of epilepsy.
| Old ILAE etiological classification7 | Proposed new ILAE etiological classification10 | ||
|---|---|---|---|
| Idiopathic | A syndrome that is only epilepsy, with no underlying structural brain lesion or other neurologic signs or symptoms. Typically age-dependent epilepsy syndromes. | Genetic | Epilepsies in which a genetic etiology has been proven, or can be deduced. Most epilepsies previously considered idiopathic are now considered genetic. |
| Symptomatic | Epilepsy in which the seizures are a result of and identifiable disease process, such as an inborn error or metabolism, CNS infection, or structural brain lesion. | Structural | Epilepsies secondary to an identifiable structural brain lesion. The lesion(s) may be congenital (e.g. lissencephaly), or acquired (e.g. traumatic), static or progressive (e.g. neoplasm). Some structural epilepsies are genetically determined (e.g. tuberous sclerosis) |
| Cryptogenic | Epilepsy in which the seizures are believed to be symptomatic, but no identifiable etiology has been identified. | Metabolic | Epilepsies secondary to inherited metabolic disease where the epilepsy is considered as a result of the metabolic derangement. |
| Immune | Epilepsies secondary to a pathological immune-mediated process, where there is evidence of CNS inflammation (e.g. autoimmune encephalitis). | ||
| Infectious | Epilepsies secondary to infection such as cerebral malaria, or viral encephalitis. | ||
| Unknown | Epilepsies in which no identifiable cause has been found, nor can be deduced. | ||
Overall, it is estimated that approximately 40–60% of childhood epilepsies have genetic etiology, and 25% have structural or metabolic etiology, whilst approximately 25% of cases remain of unknown cause.17 Increases in the sensitivity and availability of genetic, biochemical, and neuroimaging testing will result in a fall in the proportion of unknown etiology cases in future.14 The genetic causes of both metabolic and structural epilepsies are being increasingly understood, so the boundaries between these categories are becoming progressively blurred. In the future, the term “genetic” may well become outdated, to be replaced by more precise terminology.
For children in whom epilepsy presents at a young age, or is associated with developmental regression or developmental impairment, if no definitive etiological diagnosis has been made through neuroimaging, it is reasonable to consider investigation for an underlying genetic or metabolic cause. The approach to such investigation will depend on the following: the nature of the patient's epilepsy and associated comorbidities; the population background of the patient and the prevalence of specific genetic/metabolic conditions within that population; and the availability of specific diagnostic testing locally or regionally. Identifying a metabolic etiology may have significant implications for treatment approach.
ComorbiditiesThe most commonly observed comorbidities in childhood epilepsy are developmental impairments, learning disabilities, autism spectrum disorders (ASD), attention deficit hyperactivity disorder (ADHD), and behavioral problems. Given that epilepsy is a disorder affecting the brain, it does not come as a surprise that these comorbidities have a neurological basis. Recently, genetic studies have demonstrated a significant amount of genetic overlap between epilepsy and other neurodevelopmental disorders, suggesting that they all have related neurobiological determinants. Overall, it has been estimated that 80% of children with epilepsy have cognitive, psychosocial, or executive function problems.18 30% of children with epilepsy have developmental delay, 25% have language delay, 5–15% have ASD, and 20–30% have ADHD.19–22 Recognition and management of these co-morbidities is essential, since they can often be bigger priorities for the family than the epileptic seizures themselves.
The role of the EEGAs highlighted above, the EEG is a useful tool to assist the clinician in the classification of seizure types and epilepsy syndromes. However it must be emphasized that EEG should not be used as a diagnostic test for epilepsy, since it has both poor specificity and sensitivity. One-third of patients with epilepsy will have a completely normal interictal EEG,9 whilst 5% of children without epilepsy will demonstrate frank epileptiform abnormalities.23 When there is clinical doubt as to whether episodes that are occurring frequently are epileptic or not, video-telemetry or ambulatory EEG can be very useful, since most epileptic events will have an EEG correlate.
ManagementManagement of childhood epilepsy must be holistic, giving due consideration to the individual needs and expectations of the patient and their family. The concepts of “epilepsy” and “the epilepsies” are not easy to grasp, and a key role of the clinician is to help the patient and their family make sense of a condition that has traditionally been misunderstood and stigmatized. Expert management relies on having a sound knowledge of the pharmacological treatment options, including their indications, interactions, and side-effect profiles. Non-pharmacological treatment options include epilepsy surgery, the ketogenic diet, and neurostimulation techniques. Non-pharmacological treatment may allow some children whose epilepsy is refractory to medical treatment to become seizure-free, and may substantially reduce the seizure burden for others. Managing epilepsy is about more than controlling seizures, and management within a multidisciplinary team may be the optimum way to ensure that all of a child's epilepsy-related needs are met.24 Such a team may include pediatricians or pediatric neurologists, neurophysiologists, psychologists, occupational therapists, physiotherapists, speech and language therapists, specialist nurses, dieticians, radiologists, and surgeons.
Indications for starting therapyIn the Dutch study of epilepsy in childhood, after a single unprovoked epileptic seizure, 46% of children were found to have no further seizures over a 2-year follow-up period.25 Based on this high non-recurrence rate, coupled with good evidence that early AED treatment does not influence long-term seizure outcome,26 it is generally advised that treatment is not started following a single seizure.27 After two unprovoked seizures, the probability of a third event ranges from 60% to 90%,28 and therefore consideration of treatment initiation is reasonable.
Nonetheless, treatment initiation cannot be simplified to “one seizure=no treatment; more than one seizure=treatment.” The recently adopted ILAE operational definition of epilepsy (Table 1) recognizes that, in some circumstances, such as the presence of structural or metabolic etiology, the risk of recurrence after a single seizure is as high as it is after two seizures in the general population.6 Conversely, certain childhood epilepsy syndromes, particularly BCECTS and Panayiotopoulos syndrome, typically follow a course in which very few seizures are followed by complete remission, and it is reasonable to avoid treating many children with these syndromes with medication.29,30 The decision on whether to start treatment is therefore multifaceted and must bear in mind the following: age, syndromic diagnosis if applicable, etiology, and acceptability of a further seizure (which may be reduced if previous seizures have been prolonged or traumatic).
Goals of therapyThe ideal goal of AED therapy is cessation of epileptic seizures without undesirable side effects. Treating seizures is desirable, as they are frightening for families; can directly lead to medical complications, such as injury or aspiration; and may interfere with education, leisure, or employment. The term epileptic encephalopathy is used to describe epilepsies in which epileptic activity itself is believed to contribute to severe cognitive and behavioral impairments above and beyond what would be expected from the underlying pathology alone.31 Examples of epilepsies in which effective control of epileptic activity appears to go hand in hand with improved cognitive performance are WS and Landau–Kleffner syndrome, otherwise known as acquired epileptic aphasia. The relative contribution of the underlying etiology versus ongoing epileptic activity – both clinical seizures and subclinical epileptiform activity seen on EEG – to cognitive and behavioral impairment in people with epilepsy will vary in different epilepsy syndromes. Even in so-called “benign” epilepsy syndromes, many children will have specific or general cognitive impairment. The term benign is therefore likely to disappear from the epilepsy lexicon in time. Parents must be aware that controlling the seizures may not have any impact on associated learning disability.
Effectiveness of therapyOverall, approximately 60% of children achieve complete seizure freedom once they are established on a therapeutic dose their first AED. It is not currently possible to predict reliably which children will respond well to AEDs, although certain risk factors, such as age of onset <1 year, structural or metabolic etiology, developmental impairment, and high frequency of seizures before treatment initiation increase the likelihood that the epilepsy will be refractory.32,33
Evidence-based AED treatmentThere are 26 AEDs currently licensed for treatment of epilepsy in childhood. Among these medications, the older ones have attained their license through longstanding established use, and the newer ones were generally licensed after demonstrating a >50% reduction in seizure frequency when used as add-on therapy in refractory epilepsy. Meanwhile, the evidence base for which AED to use as first line treatment remains limited, with very few studies measuring long-term outcomes.
The choice of initial treatment should take into account the seizure type(s), epilepsy syndrome, age, etiology, comorbidities, and potential interactions with other medications.34
Generalized or focal seizuresThe SANAD trials used randomized unblinded methodology to compare initial treatment options, with epilepsies broadly stratified into two groups: epilepsy with focal seizures (previously termed partial epilepsy), and epilepsy with generalized seizures (previously termed generalized epilepsy).35,36 The mean age of subjects was 38.3 years in the focal group and 22.5 years in the generalized group; thus, it is difficult to say whether the findings are fully applicable to children. Treatment efficacy was determined by the proportion of patients who achieved 12-month remission 2 years after randomization.
In the focal arm of SANAD, 1721 patients were randomized to treatment with lamotrigine, carbamazepine, gabapentin, or topiramate. Both lamotrigine and carbamazepine were more efficacious than gabapentin or topiramate. Lamotrigine treatment was associated with significantly fewer side effects than carbamazepine.
In the generalized arm of SANAD, 716 patients were randomized to treatment with lamotrigine, topiramate or sodium valproate. Here sodium valproate was more efficacious than both topiramate and lamotrigine. Sodium valproate was better tolerated than topiramate, with no significant difference in tolerability between sodium valproate and lamotrigine.
Syndrome-specific AED treatmentVery few trials of initial AED treatment have been stratified by epilepsy syndrome; therefore, treatment decisions are generally guided by whether focal seizures are present or not. The exception here is CAE, for which a long-term double blinded trial compared ethosuximide, sodium valproate, and lamotrigine in a population of 453 children. Freedom from failure rates at 12 months were as follows: ethosuximide (45%), sodium valproate (44%), and lamotrigine (21%). There was statistically significant superiority of ethosuximide and sodium valproate over lamotrigine. The sodium valproate group experienced the most undesirable side effects, most notably attention dysfunction.
Another syndrome for which there is emerging evidence for specific AED treatment is WS, which is defined by the coexistence of infantile spasms and hypsarrhythmia on EEG. The syndrome starts in infancy, is frequently associated with developmental regression, and may evolve into Lennox–Gastaut syndrome. Weak evidence from retrospective cohorts has traditionally led many clinicians to use either vigabatrin or corticosteroid therapy. In a head-to head unblinded trial, there was no difference in seizure control at 12–24 months of age between both treatment options.37 It has been recently demonstrated that combined therapy with both vigabatrin and corticosteroids results in improved seizure outcomes at 12–24 months of age when compared with corticosteroid therapy alone.38
There are no other syndrome-specific AED approaches based on grade 1 or 2 clinical evidence. There are several clinical reports of patients with myoclonic seizures experiencing a deterioration in myoclonus when sodium channel-blocking medications are started. Sodium channel blockers include carbamazepine, lamotrigine, and phenytoin. These AEDs are therefore often avoided in syndromes characterized by myoclonic, seizures such as JME39 and Dravet syndrome.40
Etiology-specific therapyTreating epilepsy by etiology is an attractive concept, since targeting the underlying disease mechanism would appear to be a more specific approach. Epilepsy can be secondary to a great number of individually rare inherited metabolic diseases, and when specific treatment for these is initiated seizure control often improves. Pyridoxine dependency and GLUT1 deficiency syndrome may present with severe epilepsy that is refractory to AED therapy, but respond very well to specific treatment with pyridoxine and the ketogenic diet, respectively.
Recent advances in genetic understanding of epilepsy may in future lead to the stratification of patients with epilepsy into gene-specific AED therapy, though large cohorts will be required to build a strong evidence base. Promising results have been observed with everolimus treatment for patients with tuberous sclerosis,41 and stiripentol treatment in Dravet syndrome, a severe epileptic encephalopathy whose onset is in infancy, caused by mutations in the SCN1A gene.42
Monitoring therapyRegular monitoring of antiepileptic drug levels is rarely indicated. The best way to monitor for toxicity is to keep the patient under regular follow-up and to recommend that the family seek medical attention if they notice any potential side effects. AEDs are generally much better tolerated if they are introduced at a low dose, which is incrementally titrated upwards. Checking AED levels may be useful for certain medications with a narrow therapeutic window, such as phenytoin, phenobarbital, and triple bromide, and in situations of treatment failure and where it is suspected that subtherapeutic levels may be being achieved.
Refractory epilepsyWhen the first AED does not work, how to select the second one?If seizure freedom is not achieved with the first AED, the chance of attaining complete remission with a second AED is 40%.43 For every AED that is unsuccessful, the chances of achieving remission with a different AED diminish proportionately. Ultimately, one-third of children with epilepsy will continue to have epileptic seizures regardless of medication.44 The ILAE has set forth a practical definition of drug-resistant epilepsy as epilepsy in which two appropriately chosen and appropriately dosed AEDs have been administered without remission of seizures.45
Choice of second-line AED therapy has no evidence base. There is also no evidence that polytherapy is more efficacious than alternative monotherapy. In the first instance, most clinicians will try alternative monotherapy, although there will typically be a period of overlap whilst one AED is gradually withdrawn and another is introduced. Once two or more monotherapy approaches have been unsuccessful, a polytherapy approach may then be tried. When considering polytherapy, interactions between AEDs must be considered, as this may affect dosing. For example, the lamotrigine dose should be halved when administered concomitantly with sodium valporate due to the enzyme-inhibiting properties of the latter. It is generally recommended that the AED regimen does not include medications with the same broad mechanism of action. For example, both carbamazpeine and lamotrigine are sodium channel-blockers, so one should be cautious about using these two simultaneously.
When multiple AEDs fail to workFor children with refractory epilepsy with focal seizures, surgical treatment should be considered. In randomized trials of surgery versus additional AED therapy in carefully selected surgical candidates, the proportion of these children who become seizure-free after surgery is significantly greater than those randomized to receive an additional AED. Comprehensive preoperative evaluation involves a multidisciplinary approach to pinpoint the epileptogenic zone of the brain as the appropriate surgical target. High-resolution neuroimaging, ictal EEG, dynamic ictal radionucelotide scan (such as single photon-emission computed tomography [SPECT]), and detailed neuropsychological evaluation are all useful tools in this regard. Between 30% and 80% of pediatric patients achieve long-term seizure-freedom following epilepsy surgery46; Non-resective surgical options may be used in highly specific situations. For example, interhemispheric disconnection may be used in cases where seizures originating from one hemisphere are frequently generalizing.
Another alternative to further AED trials in refractory epilepsy is the ketogenic diet, which uses a highly specialized diet to maintain the presence of ketone bodies in the systemic circulation. Ketone bodies appear to have an anti-epileptic effect, although the exact mechanism is not well understood. The ketogenic diet appears to be equally as efficacious as add-on AED therapy. In one study 16% of children with refractory epilepsy became seizure-free on the diet.47 For some families, administration of the ketogenic diet may be challenging.
Vagus nerve stimulation (VNS) is another technique that is often used in refractory epilepsies. However, it should only be used once comprehensive surgical evaluation has excluded the possibility of targeted lesional treatment. Seizure freedom is rarely observed with VNS, and it has not been established whether VNS is any more effective than additional AED therapy.48
Specific situationsClustersAlthough there is no accepted definition of cluster seizures, it is well accepted that patients with refractory epilepsy can go through self-limited periods of significantly worsened seizure control. Such periods are often precipitated by stress, intercurrent illness, or sleep deprivation. There have been no clinical trials to inform clinicians how best to manage these situations, but many have experienced that a short course of a long-acting benzodiazepine, such as clobazam, can be effective.
Status epilepticusStatus epilepticus refers to any seizure lasting longer than 30min, or multiple seizures without restitution of normal conscious level between events. It may classified as convulsive, focal, autonomic, or absence. The recommended first line therapy is a benzodiazepine, which may be administered rectally (diazepam), orally or nasally (midazolam), or intravenously (lorazepam or diazepam). If seizures persist despite one dose of benzodiazepine, a second benzodiazepine dose may be given.49 Treatment for status epilepticus following two ineffective doses of benzodiazepine has no evidence base, and responses to therapy may be highly individual. Options include rectal paraldehyde, IV phenytoin, IV phenobarbital, IV sodium valproate, and IV levetiracetam. It is recommended that children prone to episodes of status epilepticus have their personalized treatment plans kept both by the family, and by any emergency department where they are likely to be received. For the treatment of children without personalized plans, emergency departments should have generic protocols in place to ensure prompt management and appropriate dosing.34 The differential diagnosis of status epilepticus is easy to overlook in the heat of the emergency situation, but consideration should be given to the possibility of non-epileptic status, and to increased intracranial pressure, in which decorticate posturing may mimic convulsive activity. Administration of a high dose blood pressure-lowering anticonvulsants can have significant adverse effects in cases of increased intracranial pressure.
PregnancyFor women of childbearing age with epilepsy, consideration needs to be given to the potential teratogenic effects of medication. The risk to an unborn fetus from poorly controlled epilepsy outweighs the teratogenic risk conferred from any AED, but there is now clear evidence that sodium valproate is significantly more teratogenic than the other AEDs, with 5.7–16.8% of pregnancies affected. Therefore, for patients with newly diagnosed epilepsy, if they may become pregnant in the next few years, it is advisable to commence them on a medication other than sodium valproate.50
Discontinuation of therapyFor children who have been seizure-free on medication for over 2 years on monotherapy, discontinuation of AED treatment should be considered, particularly if there is no structural or metabolic underlying etiology. In children in whom AED therapy is withdrawn after 2 years of seizure freedom, 70% will remain seizure-free for the subsequent 2 years.51
Sudden unexpected death in epilepsy and mortalityIn a Finish cohort of 245 patients with childhood-onset epilepsy followed-up over 40 years, the overall standardized mortality rate was calculated as 6.4/1000 patient-years, which is approximately 1.5 times that of the general population. The vast majority of deaths occurred in adulthood, and half were not related to epilepsy. Sudden unexpected death in epilepsy (SUDEP) accounted for 23/33 epilepsy-related deaths in this cohort. The major risk factors for increased mortality are structural or metabolic etiology, frequency of seizures, and the presence of nocturnal seizures.52 Children with uncomplicated epilepsy do not appear to be at risk of SUDEP.
Future directionsDespite major recent advances in the understanding of epilepsy etiology, and the emergence of a number of new AEDs, the proportion of childhood-onset epilepsies that are refractory to treatment has largely remained unchanged. To address this issue, there is a clear need for a more stratified approach to treatment. Traditionally, stratification has been by broad seizure type (focal or generalized), and only occasionally by epilepsy syndrome. With the emergence of genetic technology it is possible that in the future, stratification will be done by genotype. In order to build a useful evidence base, large multicenter trails are needed.
ConclusionChildhood-onset epilepsies are a heterogeneous group of disorders with a wide variety of causes and an equally wide variety of presentations. The majority of patients achieve seizure freedom with treatment on a single AED, which must be chosen appropriately. Successful management needs to be underscored by a structured approach to diagnosis, which should take a multi-axial approach. Recent advances in the understanding of epilepsy etiology and pathophysiology have resulted in a reconceptualization of the classification of epilepsies. Despite the scientific advances, and despite the development of a number of new AEDs, a significant treatment gap still exists, with a proportion of patients having refractory disease. For some carefully selected patients, epilepsy surgery can lead to remission, but successful surgery must be underpinned by comprehensive pre-surgical evaluation. A personalized medical approach using stratified treatment schemes may help to deliver better outcomes in future. In order to achieve this, global efforts must be made to establish a robust evidence base.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Zuberi SM, Symonds JD. Update on diagnosis and management of childhood epilepsies. J Pediatr (Rio J). 2015;91:S67–77.




