



To review the current evidence base for the diagnosis and management of the childhood epilepsies and to draw attention to the current gaps in this evidence base. The focus will be on therapeutic aspects. Current International League Against Epilepsy (ILAE) terminology will be described and used throughout the discussion. The review will draw attention to recent advances that have been made in both our understanding and treatment of the childhood epilepsies. Potential future directions for research and treatment options will be discussed.
SourcesOriginal articles relevant to the subject were obtained from the MedLine database using pertinent MeSH terms. Relevant papers were read and assimilated. Citation searching was used.
Summary of the findingsEpilepsy is a major cause of global disease burden. Childhood epilepsies are a heterogeneous group of conditions. A multi‐axial diagnostic approach should be taken prior to making treatment and management decisions for any individual patient. For the majority of patients, successful control of seizures can be achieved with a single medication. However, a significant minority develops refractory disease. Epilepsy surgery can provide cure for a carefully selected group of these cases.
ConclusionsThere remain significant gaps the evidence base for treatment in several areas of childhood epilepsy. Concerted multi‐center efforts should be made to try to close these gaps. A personalized medicine approach may help to reduce the proportion of refectory cases of childhood epilepsy in future.
Analisar a base de evidências atual para o diagnóstico e tratamento das epilepsias da infância e chamar a atenção para as lacunas atuais nessa base de evidências. O foco será os aspectos terapêuticos. A terminologia atual da Liga Internacional contra a Epilepsia (ILAE) será descrita e usada na discussão. A análise chamará a atenção para os recentes avanços em nosso entendimento e no tratamento das epilepsias da infância. Serão discutidas possíveis orientações futuras para as opções de pesquisa e tratamento.
Fontes de dadosTrabalhos originais relevantes para o assunto foram obtidos da base de dados MedLine com termos relevantes do MeSH. Os trabalhos relevantes foram lidos e assimilados. Foi usada pesquisa de citações.
Resumo dos dadosA epilepsia é uma das maiores causas da carga global de doenças. As epilepsias da infância representam um grupo heterogêneo de doenças. Uma abordagem multiaxial do diagnóstico deve ser feita antes da tomada de decisões de tratamento de qualquer paciente individual. Na maioria dos pacientes, o controle bem‐sucedido das crises pode ser obtido com uma única medicação. Contudo, uma minoria significativa desenvolve doença refratária. A cirurgia de epilepsia pode curar um grupo cuidadosamente selecionado desses casos.
ConclusõesAinda existem lacunas significativas na base de evidências de tratamento em diversas áreas de epilepsia da infância. Devem ser envidados esforços multicêntricos concertados para tentar fechar essas lacunas. Uma abordagem médica personalizada pode ajudar a reduzir a proporção de casos refratários de epilepsia da infância no futuro.
A epilepsia é a doença neurológica crônica mais prevalente do mundo, com uma estimativa de 65 milhões de pessoas afetadas.1 A epilepsia grave figura na quarta posição entre as maiores causas da carga global de doenças.2 As estimativas brutas de incidência de epilepsia variam de 15 a 113 em cada 100 mil pessoas por ano, dependendo da população estudada.3 A incidência de epilepsia na infância é mais do que o dobro da incidência na população adulta.4
DefiniçõesA epilepsia foi definida conceitualmente como uma “disfunção do cérebro caracterizada por uma predisposição permanente para gerar crises epilépticas”. Uma crise epiléptica é uma breve ocorrência de sinais e/ou sintomas devido à atividade neuronal anormal excessiva ou sincrônica no cérebro.5 Para facilitar a aplicação prática de epilepsia como um termo diagnóstico, a Liga Internacional contra a Epilepsia (ILAE) finalizou uma definição operacional de epilepsia em 2014 (tabela 1).6 Talvez seja mais útil conceituar a epilepsia não como uma única doença, mas como um grupo de doenças, “as epilepsias”, refletindo o reconhecimento de que as causas subjacentes de crises epilépticas são variadas e numerosas e que as manifestações clínicas da atividade neuronal anormal excessiva ou sincrônica são heterogêneas.
Definição operacional de epilepsia da ILAE de 2014.6
| Doença cerebral definida por quaisquer das seguintes condições |
| 1. No mínimo duas crises não provocadas (ou reflexivas) que ocorrem em um intervalo de 24 horas |
| 2. Uma crise não provocada (ou reflexiva) e uma probabilidade de outras crises semelhantes ao risco geral de recidiva (no mínimo 60%) após duas crises não provocadas que ocorrem nos 10 anos seguintes |
| 3. Diagnóstico de uma síndrome de epilepsia |
No que diz respeito a um diagnóstico de epilepsia, recomenda‐se uma abordagem multiaxial.7 Os seguintes eixos devem ser considerados:
- 1
O diagnóstico é de epilepsia?
- 2
Quais são os tipos de crise?
- 3
O diagnóstico de uma síndrome de epilepsia comprovada pode ser feito?
- 4
Qual é a causa subjacente da epilepsia?
- 5
Há alguma comorbidade?
A precisão do diagnóstico é fundamental, pois um diagnóstico de epilepsia pode ter implicações significativas para pacientes, suas famílias e o tratamento terapêutico. Contudo, fazer um diagnóstico de epilepsia pode ser desafiador, pois há várias doenças paroxísticas cujo histórico clínico pode ser parecido ao de uma crise epiléptica e porque não existe uma investigação de diagnóstico definitivo de epilepsia. Tradicionalmente, as taxas de diagnóstico errado de epilepsia têm sido altas. A avaliação especializada constatou que de 233 crianças encaminhadas a um centro terciário de epilepsia na Dinamarca, 87 (39%) não tinham epilepsia e, dessas, 35 (40%) já haviam iniciado o tratamento com medicamentos antiepilépticos (AEDs).8 Os diagnósticos diferenciados encontrados com mais frequência foram as crises de ausência não epilépticas (52,8%), crises psicogênicas não epilépticas (10,3%), síncope (3,4%), distonia (3,4%) e parassonias (3,4%). O diagnóstico errado de episódios não epilépticos quando o verdadeiro diagnóstico é a epilepsia aparenta ser menos comum. Em um estudo holandês com 888 crianças encaminhadas a um centro terciário com eventos paroxísticos, 19/124 (5,6%) crianças encaminhadas com vários eventos obscuros foram diagnosticadas com epilepsia.9 Acredita‐se que a avaliação inicial por um médico com conhecimento e experiência no diagnóstico da epilepsia reduz significativamente as taxas de diagnóstico errado.
Em muitos casos, o diagnóstico de um evento epiléptico ou não epiléptico pode ter como base uma descrição precisa do episódio ou episódios por parte do paciente ou da testemunha, sem necessidade de investigações adicionais. O uso generalizado de smartphones habilitados para vídeo facilitou muito o diagnóstico de eventos paroxísticos. Quando os eventos são recorrentes e há dúvidas sobre sua natureza, os pais devem ser fortemente incentivados a filmá‐los. A ILAE construiu uma excelente ferramenta on‐line,10 que os médicos podem usar para ver exemplos em vídeo de eventos epilépticos e não epilépticos.
Eventos de síncope normalmente podem ser diferenciados de crises epilépticas pela descrição do paciente sobre como ele(a) se sentiu antes do desmaio. Os sintomas prodrômicos típicos da síncope são tontura, perda da visão, náusea, transpiração e zumbido nos ouvidos. Em todos os casos ou em caso de desmaio, é aconselhável obter um eletrocardiograma de 12 derivações e a atenção específica para a mensuração do intervalo QT corrigido desde a arritmia cardíaca é uma diferença importante.11
Descrição dos tipos de criseAssim que uma clara descrição do(s) evento(s) tiver sido obtida, caso se acredite que sejam de natureza epiléptica, o médico deve tentar classificar o(s) tipo(s) da crise. A tabela 2 detalha a atual classificação da ILAE dos tipos de crise. As crises generalizadas são conceituadas como internas no início e rapidamente comprometem as redes bilaterais dentro do cérebro, ao passo que as crises focais são conceituadas como internas das redes limitadas a um hemisfério. As crises focais poderão progredir de forma a envolver ambos os hemisférios e resultar em características convulsivas bilaterais. O eletroencefalograma (EEG) é um complemento útil da classificação do tipo da crise, principalmente em epilepsias focais nas quais a atividade epileptiforme pode ajudar a identificar uma localização lobar.
Classificação da ILAE dos tipos de crise
| Crises generalizadas | |
| Tônico‐clônicas generalizadas | Aumento bilateral do tônus, seguido de espasmos rítmicos bilaterais contínuos do membro |
| Clônicas | Espasmos rítmicos bilaterais contínuos |
| Ausência | |
| Ausência típica | Início abrupto de consciência alterada. Memória de acontecimentos normalmente prejudicada. Automatismos verbais e manuais são comuns. Poderão ocorrer movimentos clônicos de partes da face. |
| Ausência atípica | Início e término menos abruptos do que na ausência típica. Pode estar associada à perda de tônus muscular ou a espasmos mioclônicos sutis. Geralmente mais longas do que as ausências típicas. Alteração variável de consciência |
| Ausência mioclônica | Espasmos mioclônicos rítmicos dos ombros e braços com abdução tônica, que resultam em elevação gradual dos braços durante a crise. Os espasmos mioclônicos normalmente são bilaterais, mas podem ser unilaterais ou assimétricos. Duram 10‐60 segundos. Alteração variável de consciência. |
| Ausência com mioclonia palpebral | Crises de ausência acompanhadas de espasmos mioclônicos breves, repetitivos, normalmente rítmicos e rápidos das pálpebras com o desvio simultâneo dos globos oculares para cima e extensão da cabeça. As crises normalmente são muito breves (< 6 s de duração) e várias crises ocorrem diariamente. A maior parte da consciência é mantida. |
| Tônica | Aumento bilateral do tônus dos membros que pode durar de segundos a um minuto. Normalmente ocorre nas horas acordadas e em episódios de intensidade variável. O indivíduo não está consciente durante esses episódios. Os indivíduos poderão fazer um som de expiração durante o acometimento. Pode causar ataques de queda. |
| Atônica | Perda ou diminuição repentina do tônus muscular sem características mioclônicas ou tônicas anteriores evidentes. Muito breve (< 2 segundos) e pode envolver a cabeça, o tronco ou os membros. |
| Mioclônica | |
| Mioclônica | Espasmos (breves contrações musculares) isolados ou em série. Cada espasmo normalmente dura milissegundos. |
| Mioclonia negativa | Breve cessação do tônus muscular de fundo, com duração de menos de 500 milissegundos. O movimento consequente produzido pode ter dois componentes, uma perda inicial de postura causada pela mioclonia negativa e por um movimento voluntário e compensatório para restaurar a postura. Pode ocorrer isoladamente ou em série. |
| Mioclônica atônica | Crise mioclônica seguida de uma crise atônica. Às vezes, uma série de espasmos mioclônicos ocorre antes da atonia. A cabeça e os membros são afetados e normalmente resultam em uma queda rápida. |
| Mioclônica tônica | Crise mioclônica seguida de uma crise tônica. Às vezes, uma série de espasmos mioclônicos ocorre antes do aumento do tônus. |
| Crises focais. Podem ser classificadas de acordo com as características e/ou a lateralidade e/ou a localização lobar | |
| De acordo com as características (clínicas). Um ou mais tipos de características podem estar presentes durante qualquer crise focal. | |
| Aura | As auras são subjetivas e poderão ser sensoriais ou experienciais. Uma aura sensorial envolve uma sensação sem um sinal clínico objetivo, que pode ser visual, auditiva, olfativa, epigástrica etc. Uma aura experiencial envolve fenômenos subjetivos afetivos, mnemônicos ou perceptivos, incluindo episódios de despersonalização e alucinação. |
| Motoras | Uma característica motora envolve a atividade motora e poderá consistir em um aumento (positiva) ou redução (negativa) da contração muscular. |
| Autonômicas | Caracterizadas por fenômenos autonômicos, que podem envolver funções cardiovasculares, gastrointestinais, vasomotoras e termorreguladoras. Os exemplos incluem palpitações, náusea, frio na barriga, fome, dor no peito, vontade de urinar ou defecar, arrepios, sensações sexuais, sensação de calor ou frio, piloereção, palidez, taquicardia ou bradicardia/assistolia, vermelhidão, alterações na pupila e lacrimejamento. |
| Discognitivas | Envolve alteração na consciência ou na responsividade. |
| De acordo com a localização hemisférica (clínica ou eletrofisiológica) | |
| DireitaEsquerda | As indicações da localização hemisférica podem ser sutis, principalmente em crises que envolvem rapidamente redes bilaterais. Cabeça virada, desvio ocular e movimentos clônicos unilaterais, todos lateralizam para o hemisfério contralateral. |
| De acordo com a localização lobar (clínica ou eletrofisiológica) | |
| Frontal | As crises do lobo frontal normalmente são breves. As características motoras são destacadas. Pode haver vocalização, comportamento bizarro, desvio da cabeça e dos olhos e incontinência urinária. |
| Temporal | Caracterizado por inibição comportamental e características discognitivas. Automatismos e auras são comuns. |
| Parietal | As características ictais normalmente são subjetivas e incluem características sensoriais positivas e/ou negativas, como parestesia, desorientação e alucinações visuais complexas. |
| Occipital | Caracterizado por aura visual, formas tipicamente multicoloridas. |
| Espasmos epilépticos | |
| Flexão repentina, extensão ou flexoextensão de músculos proximais e tronculares com duração de 1‐2 segundos, ou seja, mais do que um espasmo mioclônico (que dura milissegundos), mas não tanto quanto uma crise tônica (que dura>2 segundos). Os espasmos ocorrem geralmente em uma série, normalmente ao acordar. Formas sutis poderão ocorrer com somente movimento do queixo, caretas ou acenos de cabeça. Os espasmos podem ser bilateralmente simétricos, assimétricos ou unilaterais. | |
Por meio da assimilação das informações clínicas sobre a epilepsia de um paciente, incluindo tipos de crise, idade de acometimento e comorbidades associadas, pode ser possível diagnosticar uma síndrome de epilepsia específica. Muitas síndromes de epilepsia são associadas a achados específicos sobre EEG, portanto obter um EEG é muito útil para a classificação das síndromes. É importante conseguir um diagnóstico da síndrome, já que ele pode fundamentar decisões terapêuticas, investigações adicionais e prognóstico. O website da ILAE fornece as características clínicas e de EEG de 31 síndromes de epilepsia.10 As estimativas da proporção de casos de epilepsia da infância que podem ser diagnosticados com uma síndrome de epilepsia variaram entre 15,7% e 37,1%, dependendo da população incluída.12‐14 As síndromes infantis de epilepsia mais prevalentes parecem ser a epilepsia benigna da infância com espículas centrotemporais (BCECTS), epilepsia ausência da infância (EAI), epilepsia ausência juvenil (EAJ), epilepsia mioclônica juvenil (EMJ) e síndrome de West (SW). Muitas das epilepsias que não se encaixam em um diagnóstico sindrômico ainda podem ser descritas em termos de tipos de crise e etiologia (tabela 3).
As síndromes de epilepsia da infância encontradas com mais frequência
| Síndrome de epilepsia | Proporção total de epilepsia da infância | Idade de acometimento | Descrição da síndrome |
|---|---|---|---|
| Síndrome de West15 | 2‐3% | 3‐12 meses | Espasmos infantis com hipsarritmia no EEG. Normalmente lesões cerebrais secundárias a estruturais. Normalmente é observado o comprometimento global do desenvolvimento. |
| Epilepsia ausência da infância15 | 2‐5% | 8‐12 anos | Múltiplas crises ausência por dia em uma criança de outra forma saudável. As ausências podem ser invariavelmente provocadas pela hiperventilação e são associadas a descargas de picos de ondas generalizadas de 2,5‐3,5Hz no EEG |
| Epilepsia benigna da infância com espículas centrotemporais15 | 3‐8% | 3‐14 anos | Breves crises hemifaciais que podem ser secundariamente generalizadas se ocorrerem no período noturno. O EEG é característico, mostra um histórico normal com ondas acentuadas de elevada amplitude nas regiões centrotemporais, ativadas pela sonolência e pelo sono; 98% das crianças ficam livres das crises sem medicamento até os 18 anos. |
| Epilepsia ausência jvenil15 | 1‐4% | 8‐20 anos | Crises de ausência típicas relativamente não frequentes, bem como crises convulsivas generalizadas em uma criança ou jovem adulto de outra forma normal. As descargas epileptiformes generalizadas são vistas no EEG interictal. |
| Epilepsia mioclônica juvenil15 | 5‐11% | 8‐25 anos | Crises mioclônicas frequentes mais proeminentes de manhã em uma criança ou jovem adulto de outra forma normal. A maioria dos pacientes também tem crises convulsivas generalizadas. As descargas epileptiformes generalizadas são vistas no EEG interictal. |
Na maioria das epilepsias, a investigação de uma etiologia subjacente é garantida, independentemente de um diagnóstico de síndrome de epilepsia ter sido feito ou não. As exceções a essa regra são casos típicos de BCECTS, EAI, EAJ e EMJ. Embora todas essas epilepsias tenham uma etiologia principalmente genética, a investigação genética não é, hoje, útil clinicamente, pois diversos genes de susceptibilidade estão envolvidos e não há correlação estabelecida entre achados genéticos e o prognóstico ou a administração terapêutica. Essas mesmas síndromes são muito raramente associadas a anomalias estruturais do cérebro, de forma que a neuroimagiologia também não é necessária.15 Em todos os outros casos de epilepsia da infância, incluindo casos atípicos das síndromes acima, é indicada a neuroimagiologia. A finalidade da neuroimagiologia é identificar qualquer lesão cerebral estrutural causal. Algumas lesões, como neoplasias, podem exigir tratamento imediato, ao passo que outras, como displasia cortical focal, podem ser tratadas por cirurgia em um momento posterior. Neuroimagens anormais são encontradas em 50% dos novos casos de epilepsia da infância em que existem crises focais e em 15‐20% dos casos as neuroimagens fornecem informações úteis sobre a etiologia e/ou o foco da crise.16 A ressonância magnética é a modalidade de imagiologia escolhida.
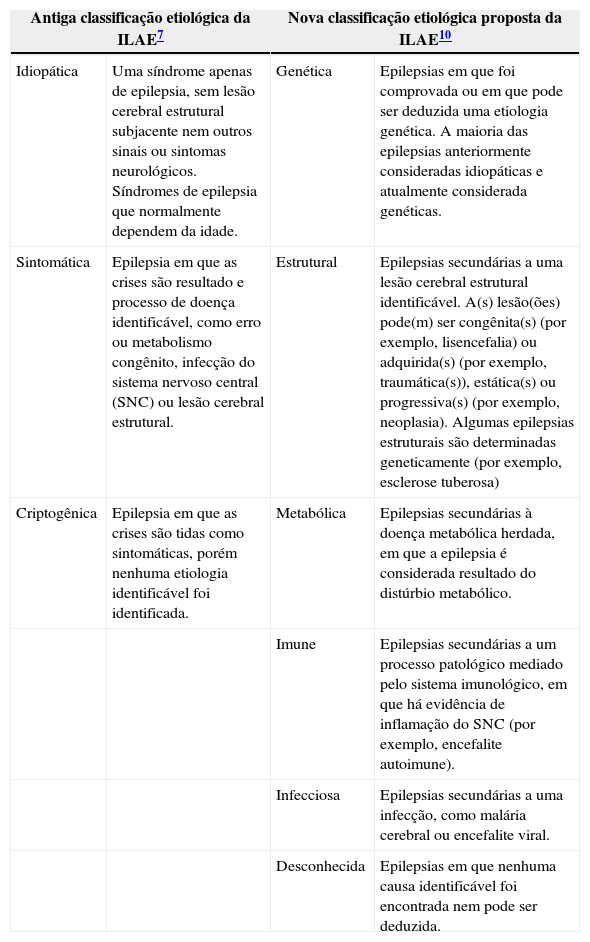
A primeira tentativa significativa de reclassificação das epilepsias em linha com o entendimento moderno da doença e de seus mecanismos foi feita pela ILAE em 2010. As principais alterações na nomenclatura oficial da ILAE focaram na categorização etiológica. Os termos “idiopática”, “sintomática” e “criptogênica” foram substituídos pelos termos mais precisos “genética”, “estrutural” e “metabólica”. A inclusão das outras categorias etiológicas “imune” e “infecciosa” se encontra atualmente em discussão (tabela 4).
Classificação etiológica da epilepsia da ILAE
| Antiga classificação etiológica da ILAE7 | Nova classificação etiológica proposta da ILAE10 | ||
|---|---|---|---|
| Idiopática | Uma síndrome apenas de epilepsia, sem lesão cerebral estrutural subjacente nem outros sinais ou sintomas neurológicos. Síndromes de epilepsia que normalmente dependem da idade. | Genética | Epilepsias em que foi comprovada ou em que pode ser deduzida uma etiologia genética. A maioria das epilepsias anteriormente consideradas idiopáticas e atualmente considerada genéticas. |
| Sintomática | Epilepsia em que as crises são resultado e processo de doença identificável, como erro ou metabolismo congênito, infecção do sistema nervoso central (SNC) ou lesão cerebral estrutural. | Estrutural | Epilepsias secundárias a uma lesão cerebral estrutural identificável. A(s) lesão(ões) pode(m) ser congênita(s) (por exemplo, lisencefalia) ou adquirida(s) (por exemplo, traumática(s)), estática(s) ou progressiva(s) (por exemplo, neoplasia). Algumas epilepsias estruturais são determinadas geneticamente (por exemplo, esclerose tuberosa) |
| Criptogênica | Epilepsia em que as crises são tidas como sintomáticas, porém nenhuma etiologia identificável foi identificada. | Metabólica | Epilepsias secundárias à doença metabólica herdada, em que a epilepsia é considerada resultado do distúrbio metabólico. |
| Imune | Epilepsias secundárias a um processo patológico mediado pelo sistema imunológico, em que há evidência de inflamação do SNC (por exemplo, encefalite autoimune). | ||
| Infecciosa | Epilepsias secundárias a uma infecção, como malária cerebral ou encefalite viral. | ||
| Desconhecida | Epilepsias em que nenhuma causa identificável foi encontrada nem pode ser deduzida. | ||
Em geral, estima‐se que cerca de 40‐60% das epilepsias da infância têm etiologia genética e 25% têm etiologia estrutural ou metabólica, ao passo que aproximadamente 25% dos casos continuam sendo de causa desconhecida.17 Os aumentos na sensibilidade e disponibilidade de exames genéticos, bioquímicos e de neuroimagiologia resultarão em uma queda na proporção de casos de etiologia desconhecida no futuro.14 Cada vez mais entendemos as causas genéticas das epilepsias metabólica e estrutural. Dessa forma, os limites entre essas categorias estão se tornando progressivamente confusos. No futuro, o termo “genética” pode se tornar bem ultrapassado e deve ser substituído por uma terminologia mais precisa.
Com relação a crianças em que a epilepsia se apresenta muito cedo ou está associada a uma regressão ou a um comprometimento do desenvolvimento, se nenhum diagnóstico etiológico definitivo tiver sido feito por meio da neuroimagiologia, é razoável considerar a investigação de uma causa genética ou metabólica subjacente. A abordagem a essa investigação dependerá do seguinte: a natureza da epilepsia do paciente e as comorbidades correspondentes; o histórico populacional do paciente e a prevalência de doenças genéticas/metabólicas específicas nessa população; e a disponibilidade de exames de diagnóstico específicos local ou regionalmente. A identificação de uma etiologia metabólica poderá ter implicações significativas na abordagem do tratamento.
ComorbidadesAs comorbidades mais comumente encontradas na epilepsia da infância são comprometimentos no desenvolvimento, dificuldades de aprendizagem, transtornos do espectro autista (TEA), transtorno do déficit de atenção com hiperatividade (TDAH) e problemas comportamentais. Considerando que a epilepsia é uma doença que afeta o cérebro, não é surpresa que essas comorbidades tenham uma base neurológica. Recentemente, estudos genéticos demonstraram uma quantidade significativa de sobreposição genética entre epilepsia e outras disfunções no neurodesenvolvimento. Isso sugere que todas elas têm determinantes neurobiológicos relacionados. De modo geral, foi estimado que 80% das crianças com epilepsia apresentam problemas cognitivos, psicossociais ou de função executiva;18 30% das crianças com epilepsia apresentam atraso de desenvolvimento, 25% apresentam atraso de linguagem, 5‐15% apresentam TEA e 20‐30% apresentam TDAH.19‐22 É essencial reconhecer e tratar essas comorbidades, pois, normalmente, elas podem ser maiores prioridades para a família do que as próprias crises epilépticas.
O papel do EEGConforme destacado anteriormente, o EEG é uma ferramenta útil para auxiliar o clínico na classificação dos tipos de crise e síndromes de epilepsia. Contudo, deve ser enfatizado que o EEG não deve ser usado como exame de diagnóstico para epilepsia, já que tem especificidade e sensibilidade baixas. Um terço dos pacientes com epilepsia apresentarão um EEG interictal9 completamente normal, ao passo que 5% das crianças sem epilepsia demonstrarão anormalidades epileptiformes francas.23 Quando há dúvida clínica se episódios que ocorrem com mais frequência são epilépticos ou não, videotelemetria ou EEG ambulatório podem ser muito úteis, já que a maioria dos casos epilépticos terá um EEG correlacionado.
TratamentoO tratamento da epilepsia da infância deve ter uma abordagem holística, considerando devidamente as necessidades e expectativas individuais do paciente e de sua família. Os conceitos de “epilepsia” e “as epilepsias” não são fáceis de entender e um papel importante do médico é ajudar o paciente e sua família a entender uma doença tradicionalmente mal entendida e estigmatizada. O tratamento especializado tem como base um conhecimento sólido das opções de tratamento farmacológico, incluindo indicações, interações e efeitos colaterais. Ter acesso a opções de tratamento não farmacológico, incluindo cirurgia de epilepsia, dieta cetogênica e técnicas de neuroestimulação, pode permitir que algumas crianças cuja epilepsia seja refratária ao tratamento medicamentoso fiquem livres das crises e pode reduzir substancialmente a quantidade de crises em outras pessoas. Tratar a epilepsia diz mais respeito a controlar as crises e a gestão de uma equipe multidisciplinar pode ser a forma ideal de garantir que todas as necessidades relacionadas à epilepsia de uma criança sejam atendidas.24 Uma equipe dessas pode incluir pediatras ou neurologistas pediátricos, neurofisiologistas, psicologistas, terapeutas ocupacionais, fisioterapeutas, fonoaudiólogos, enfermeiras especializadas, radiologistas e cirurgiões.
Indicações para o início da terapiaNo estudo holandês da epilepsia na infância, foi observado que, após uma única crise epiléptica não provocada, 46% das crianças não tiveram outra crise em um período de acompanhamento de dois anos.25 Com base nessa alta taxa de não recidiva, aliada à boa comprovação de que o tratamento antecipado com AEDs não influencia o resultado de longo prazo das crises,26 normalmente aconselha‐se que o tratamento não seja iniciado após uma única crise.27 Após duas crises não provocadas, a probabilidade de uma terceira é de 60‐90%28 e, portanto, é razoável considerar o início do tratamento.
Contudo, o início da terapia não pode ser simplificado a “uma crise=sem tratamento/mais de uma crise=tratamento”. A definição operacional de epilepsia da ILAE recentemente adotada (tabela 1) reconhece que, em algumas circunstâncias, por exemplo, a presença da etiologia estrutural ou metabólica, o risco de recidiva após uma única crise é tão alto quanto após duas crises na população como um todo.6 Por outro lado, algumas síndromes de epilepsia da infância, principalmente a BCECTS e a síndrome de Panayiotopoulos, normalmente seguem um curso no qual muito poucas crises são seguidas da remissão completa e é razoável evitar o tratamento de muitas crianças com essas síndromes com medicamentos.29,30 A decisão de iniciar o tratamento é, portanto, multifacetada e deve‐se ter em mente as seguintes características: idade; diagnóstico sindrômico, se aplicável; etiologia; e aceitabilidade de uma crise posterior (que pode ser reduzida caso crises anteriores tenham sido prolongadas ou traumáticas).
Objetivos da terapiaO objetivo ideal da terapia com AEDs é causar a cessação das crises epilépticas sem causar efeitos colaterais indesejáveis. O tratamento das crises é desejável porque as crises epilépticas são assustadoras para as famílias, podem levar a complicações médicas, como lesão ou aspiração, diretamente e podem interferir na educação, no lazer ou no trabalho. O termo encefalopatia epiléptica é usado para descrever epilepsias nas quais se acredita que a própria atividade epiléptica contribui para deficiências cognitivas e comportamentais graves acima e além do esperado para a patologia subjacente sozinha.31 Exemplos de epilepsias nas quais o controle efetivo da atividade epiléptica parece associado com a melhoria do desempenho cognitivo são a síndrome de West e a síndrome de Landau‐Kleffner, também conhecida como afasia epiléptica adquirida. A contribuição relativa da etiologia subjacente em comparação com a atividade epiléptica contínua – tanto as crises clínicas quanto a atividade epileptiforme vista no EEG – para a deficiência cognitiva e comportamental em pessoas com epilepsia variará em diferentes síndromes de epilepsia. Mesmo nas síndromes de epilepsia chamadas “benignas”, muitas crianças terão deficiências cognitivas específicas ou gerais. O termo “benignas”, portanto, provavelmente desaparecerá do léxico associado à epilepsia em algum tempo. Os pais devem estar cientes de que o controle das crises pode não exercer mpacto sobre a dificuldade de aprendizagem correspondente.
Eficácia da terapiaEm geral, cerca de 60% das crianças chegam a ficar completamente livres de crises assim que recebem sua primeira dose terapêutica do medicamento antiepiléptico (AED). Atualmente, não é possível prever, de forma confiável, quais crianças responderão bem aos AEDs, embora certos fatores de risco, como a idade de acometimento<1 ano, a etiologia estrutural ou metabólica, o comprometimento do desenvolvimento e a elevada frequência de crises antes do início do tratamento, aumentem a probabilidade de a epilepsia ser refratária.32,33
Tratamento com AEDs com base nas evidênciasExistem 26 AEDs atualmente licenciados para o tratamento da epilepsia da infância. Dentre esses medicamentos, os mais antigos obtiveram sua licença por meio do uso de longa data estabelecido e os mais recentes normalmente obtiveram sua licença demonstrando uma redução de>50% na frequência de crises quando usados como terapia adicional da epilepsia refratária. Enquanto isso, a base de evidências com relação à qual o AED deve ser usado como tratamento de primeira linha continua limitada, com muito poucos estudos que medem os resultados de longo prazo.
Recomenda‐se que a escolha do tratamento inicial deve levar em consideração o(s) tipo(s) de crise, a síndrome de epilepsia, a idade, a etiologia, as comorbidades e possíveis interações com outros medicamentos.34
Crises generalizadas ou focaisOs ensaios clínicos do Standard And New Antiepileptic Drugs (Sanad) usaram a metodologia não cega randomizada para comparar as opções de tratamento inicial com as epilepsias amplamente estratificadas em dois grupos: epilepsia com crises focais (anteriormente denominada epilepsia parcial) e epilepsia com crises generalizadas (anteriormente denominada epilepsia generalizada).35,36 A idade média dos indivíduos era de 38,3 anos no grupo focal e 22,5 anos no grupo generalizado. Assim, é difícil dizer se os achados são totalmente aplicáveis a crianças. A eficácia do tratamento foi determinada pela proporção de pacientes que atingiram a remissão de 12 meses dois anos depois da randomização.
No segmento focal do Sanad, 1.721 pacientes foram randomizados ao tratamento com lamotrigina, carbamazepina, gabapentina ou topiramato. A lamotrigina e a carbamazepina foram mais eficazes do que a gabapentina ou o topiramato. O tratamento com lamotrigina foi associado a efeitos colaterais significativamente menores do que com a carbamazepina.
No grupo generalizado do Sanad, 716 pacientes foram randomizados ao tratamento com lamotrigina, topiramato ou valproato de sódio. O valproato de sódio foi mais eficaz do que o topiramato e a lamotrigina. O valproato de sódio foi mais bem tolerado do que o topiramato, sem diferença significativa na tolerância entre o valproato de sódio e a lamotrigina.
Tratamento com AEDs específicos da síndromeMuito poucos ensaios clínicos de tratamento inicial com AEDs foram estratificados por síndrome de epilepsia. Assim, as decisões de tratamento normalmente são norteadas pela presença ou ausência de crises focais. A exceção é a EAI, com relação à qual um estudo duplo cego de longo prazo foi feito e comparou etossuximida, valproato de sódio e lamotrigina em uma população de 453 crianças. As taxas de liberdade decorrentes de falha em 12 meses eram as seguintes: etossuximida (45%), valproato de sódio (44%), lamotrigina (21%). Houve superioridade estatisticamente significativa de etossuximida e de valproato de sódio em relação à lamotrigina. O grupo de valproato de sódio apresentou os efeitos colaterais mais indesejáveis, sobretudo disfunção de atenção.
Outra síndrome para a qual existem evidências emergentes de tratamento específico com AEDs é a síndrome de West. A síndrome de West é definida pela coexistência de espasmos infantis e hipsarritmia no EEG. A síndrome começa na infância, é frequentemente associada à regressão de desenvolvimento e pode evoluir para a síndrome de Lennox‐Gastaut. Uma evidência fraca a partir de coortes retrospectivas leva tradicionalmente muitos médicos a usar vigabatrina ou terapia com corticosteroides. Em um estudo não cego de comparação direta, não havia diferença no controle de crise em 12‐24 meses de idade entre essas duas opções de tratamento.37 Recentemente, foi demonstrado que a terapia combinada com vigabatrina e corticosteroides resulta em melhoria nos resultados de crise em 12‐24 meses de idade em comparação com a terapia apenas com corticosteroides.38
Não há outra abordagem de AEDs específicos da síndrome com base em evidências clínicas da série 1 ou 2. Há diversos relatos clínicos de pacientes com crises mioclônicas que passam por um agravamento da mioclonia quando os medicamentos que bloqueiam os canais de sódio começam a ser administrados. Entre os bloqueadores dos canais de sódio estão incluídas a carbamazepina, a lamotrigina e a fenitoína. Esses AEDs são, portanto, normalmente evitados em síndromes caracterizadas por crises mioclônicas, como a EMJ39 e a síndrome de Dravet.40
Terapia específica de etiologiaO tratamento da epilepsia por meio da etiologia é um conceito atrativo, pois focar no mecanismo da doença de base parece ser uma abordagem mais específica. A epilepsia pode ser uma doença secundária relacionada a várias doenças metabólicas individualmente raras, porém conjuntamente herdadas, e quando o tratamento específico para essas doenças é iniciado normalmente o controle das crises melhora. As síndromes de dependência de piridoxina e deficiência de GLUT1 podem apresentar epilepsia grave refratária à terapia com AEDs, porém os pacientes respondem muito bem ao tratamento específico com pridoxina e à dieta cetogênica, respectivamente.
Avanços recentes no entendimento genético da epilepsia poderão, no futuro, fazer com que os pacientes com epilepsia sejam estratificados de acordo com a terapia com AEDs específica do gene, embora coortes grandes sejam necessárias para construir uma forte base de evidências. Foram observados resultados promissores no tratamento com everolimo em pacientes com esclerose tuberosa41 e no tratamento com estiripentol na síndrome de Dravet, grave encefalopatia epiléptica acometida na infância, causada por mutações no gene SCN1A.42
Terapia de monitoramentoO monitoramento regular dos níveis de medicamento antiepiléptico é raramente indicado. A melhor maneira de monitorar a toxicidade é manter o paciente sob análise regular e recomendar que a família busque atendimento médico caso observe quaisquer possíveis efeitos colaterais. Os AEDs normalmente são muito mais bem tolerados se introduzidos a uma dose baixa, gradativamente ajustada para cima. Verificar os níveis de AEDs pode ser útil com relação a alguns medicamentos com janelas terapêuticas estreitas, como a fenitoína, o fenobarbital e o elixir triplo de brometo, e em situações em que houver falha no tratamento e suspeita de que os níveis subterapêuticos podem estar sendo atingidos.
Epilepsia refratáriaQuando o 1° AED não funciona, como escolho um segundo?Se a cessação das crises não é obtida com o primeiro AED, a chance de atingir a remissão completa com um segundo AED é de 40%.43 Para cada AED malsucedido, as chances de atingir a remissão com um AED diferente diminuem proporcionalmente. Por fim, um terço das crianças com epilepsia continuará a apresentar crises epiléticas independentemente de qualquer medicamento testado.44 A ILAE elaborou uma definição prática de epilepsia resistente ao medicamento como a epilepsia em que dois AEDs adequadamente escolhidos e dosados foram usados sem a remissão das crises.45
A escolha da terapia com AED de segunda linha não tem base de evidências. Além disso, não há vidência de que a politerapia é mais eficaz do que a monoterapia opcional. Em um primeiro momento, a maioria dos médicos tentará a monoterapia opcional, embora, normalmente, haja um período de sobreposição quando um AED é retirado gradualmente e outro, introduzido. Assim que duas ou mais abordagens de monoterapia tiverem sido malsucedidas, uma abordagem de politerapia poderá então ser testada. Ao se considerar a politerapia, as interações entre os AEDs devem ser consideradas, já que isso pode afetar a dosagem. Por exemplo, a dose de lamotrigina deve ser reduzida pela metade ao ser administrada simultaneamente com o valproato de sódio devido às propriedades de inibição enzimática do valproato de sódio. Normalmente, recomenda‐se que o regime de AEDs não inclua medicamentos com o mesmo amplo mecanismo de ação. Por exemplo, a carbamazepina e a lamotrigina são bloqueadores dos canais de sódio. Dessa forma, deve‐se ter cuidado com relação ao uso combinado desses dois medicamentos.
Quando vários AEDs não funcionamCom relação a crianças com epilepsia refratária com crises focais, recomenda‐se a consideração de um tratamento cirúrgico. Nos ensaios clínicos randomizados sobre a cirurgia em comparação com a terapia adicional com AEDs em candidatos cirúrgicos selecionados com cuidado, a proporção dessas crianças que se libertaram das crises após a cirurgia é significativamente superior às randomizadas para receber um AED adicional. A avaliação pré‐operatória abrangente envolve uma abordagem multidisciplinar para apontar precisamente a zona epileptogênica do cérebro como o alvo cirúrgico adequado. A neuroimagiologia de alta resolução, o EEG ictal, a verificação dinâmica do radionuclídeo ictal, como a tomografia computadorizada por emissão de fóton único (Spect), e uma avaliação neuropsicológica detalhada são todos ferramentas úteis a esse respeito. Entre 30 e 80% dos pacientes pediátricos se libertam das crises de longo prazo após a cirurgia da epilepsia.46 As opções cirúrgicas não ressectivas poderão ser usadas em situações altamente específicas, por exemplo, a desconexão inter‐hemisférica poderá ser usada como paliativo nos casos em que as crises originárias em um hemisfério sejam frequentemente generalizantes.
Outra opção para outros ensaios clínicos com AEDs na epilepsia refratária é a dieta cetogênica, que usa uma dieta altamente especializada para manter a presença de corpos cetônicos na circulação sistêmica. Os corpos cetônicos aparentam ter um efeito antiepiléptico, embora o mecanismo exato desse efeito não seja bem entendido. A dieta cetogênica aparenta ser tão eficaz quanto a terapia adicional com AEDs. Em um estudo, 16% das crianças com epilepsia refratária40 se libertaram das crises com a dieta.47 Para algumas famílias, a administração da dieta cetogênica poderá ser desafiadora.
A estimulação do nervo vago (ENV) é outra técnica frequentemente usada em epilepsias refratárias, embora somente deva ser usada assim que a avaliação cirúrgica abrangente tiver excluído a possibilidade do tratamento da lesão alvo. A liberdade das crises raramente é vista com a ENV e não foi estabelecido se a ENV é mais eficaz do que a terapia adicional com AEDs.48
Situações específicasRepetiçõesEmbora não haja uma definição aceita de repetições de crises, reconhece‐se bem que pacientes com epilepsia refratária podem passar por períodos autolimitados de controle da crise significativamente piorado. Esses períodos normalmente são precedidos de estresse, doenças intercorrentes ou privação do sono. Não existem ensaios clínicos para informar os médicos sobre como tratar melhor essas situações, porém muitos perceberam que um curto período de uso de uma benzodiazepina de ação prolongada como o clobazam pode ser eficaz.
Estado de mal epilépticoO estado de mal epiléptico refere‐se a qualquer crise com duração maior do que 30 minutos ou múltiplas crises sem a restituição do nível normal de consciência entre episódios. O estado pode ser convulsivo, focal, autonômico ou ausente. A terapia inicial recomendada é com benzodiazepina, que pode ser administrada por via retal (diazepam), oral ou nasal (midazolam) ou intravenosa (lorazepam ou diazepam). Caso a crise persista apesar da dose de benzodiazepina, uma segunda dose de benzodiazepina poderá ser administrada.49 O tratamento para o estado de mal epiléptico após duas doses ineficazes de benzodiazepina não apresenta uma base de evidências a serem adotadas e as respostas à terapia poderão ser altamente individuais. As opções incluem paraldeído por via retal, fenitoína intravenosa, fenobarbital intravenoso, valproato de sódio intravenoso e levetiracetam intravenoso. Recomenda‐se que crianças propensas a episódios de estado de mal epiléptico tenham planos de tratamento personalizado mantidos tanto pela família quanto por qualquer departamento de emergência em que provavelmente serão recebidas. No tratamento de crianças sem planos personalizados, os serviços de emergência devem ter protocolos genéricos em vigor para garantir tratamento imediato e dosagens adequadas.34 O diagnóstico diferenciado de estado de mal epiléptico é facilmente ignorado no calor da situação de emergência, porém deve‐se considerar a possibilidade de estados não epilépticos e o aumento de pressão intracraniana, quando a postura de decorticação pode imitar a atividade convulsiva. A administração de altas doses de anticonvulsivantes para redução da pressão sanguínea pode ter efeitos adversos significativos em casos de aumento da pressão intracraniana.
GravidezEm mulheres com potencial de gravidez com epilepsia, devem‐se considerar os possíveis efeitos teratogênicos da medicação. O risco para um feto em uma gestante com epilepsia mal controlada supera o risco teratogênico conferido de qualquer AED, porém existe agora uma evidência clara de que o valproato de sódio é significativamente mais teratogênico do que outros AEDs, com 5,7‐16,8% de gestações afetadas. Portanto, com relação a pacientes com epilepsia recém‐diagnosticada, caso seja considerado possível que engravidem nos próximos anos, é aconselhável iniciar uma medicação diferente do valproato de sódio.50
Interrupção da terapiaCom relação a crianças que se libertaram das crises tomando uma medicação por>2 anos na monoterapia, a interrupção do tratamento com AEDs deve ser considerada, principalmente se não houver etiologia estrutural ou metabólica subjacente. Com relação a crianças nas quais a terapia com AEDs é interrompida depois de dois anos de libertação das crises, 70% continuarão livres das crises pelos dois anos seguintes.51
Sudep e mortalidadeEm uma coorte finlandesa de 245 pacientes com epilepsia da infância acompanhados por mais de 40 anos, a taxa de mortalidade geral padronizada foi calculada em 6,4/1.000 pacientes ao ano, aproximadamente 1,5 vez a população geral. A grande maioria dos óbitos ocorreu na vida adulta e metade não foi relacionada à epilepsia. A taxa de morte súbita inesperada em epilepsia (Sudep) representou 23/33 óbitos relacionados à epilepsia nessa coorte. Os principais fatores de risco da maior mortalidade são a etiologia estrutural ou metabólica, a frequência das crises e a presença de crises noturnas.52 As crianças com epilepsia simples não parecem correr risco de Sudep.
Futuras orientaçõesApesar dos principais avanços recentes em nosso entendimento da etiologia da epilepsia e do surgimento de vários novos AEDs, a proporção de epilepsias da infância refratárias continuou, em grande parte, inalterada. Para resolver isso, há uma clara necessidade de uma abordagem mais estratificada ao tratamento. Tradicionalmente, a estratificação tem sido feita por um amplo tipo de crise (focal ou generalizada) e somente ocasionalmente pela síndrome de epilepsia. Com o surgimento da tecnologia genética, existe a possibilidade de que, no futuro, a estratificação será possível pelo genótipo. A fim de construir uma base de evidências útil, grandes ensaios multicêntricos precisarão ser feitos.
ConclusãoAs epilepsias da infância são um grupo heterogêneo de doenças com uma grande variedade de causas e uma variedade igualmente grande de apresentações. A maioria dos pacientes se livra das crises com tratamento com base em um único AED, porém ele deve ser escolhido adequadamente. O tratamento bem‐sucedido precisa ser destacado por uma abordagem estruturada ao diagnóstico, que deve ser uma abordagem multiaxial. Os recentes avanços em nosso entendimento da etiologia e da fisiopatologia da epilepsia resultaram em uma reconceitualização de como classificamos as epilepsias. Apesar dos avanços científicos e do número de novos AEDs que estão entrando no mercado, ainda há uma lacuna significativa no tratamento, com uma proporção de pacientes com doença refratária. No que diz respeito a alguns pacientes cuidadosamente selecionados, a cirurgia pode levar a remissão, porém uma cirurgia bem‐sucedida pode ter como base uma avaliação pré‐cirúrgica abrangente. Uma abordagem personalizada aos medicamentos com o uso de esquemas estratificados de tratamento pode ajudar a gerar melhores resultados no futuro, mas, para isso, devem ser envidados esforços globais para formar uma forte base de evidências.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Zuberi SM, Symonds JD. Update on diagnosis and management of childhood epilepsies. J Pediatr (Rio J). 2015;91:S67–77.




