






The abnormalities of the genitourinary tract development are the leading cause of chronic kidney disease (CKD) in children. The diagnosis of this disease in Brazil is late and incomplete, which results in increased morbidity and mortality in this age group. Early diagnosis of this condition is the prerogative of generalist pediatricians, and the aim of this study was to review the clinical signs and symptoms associated with developmental abnormalities of the genitourinary tract.
Data sourcesBased on the description of a symbolic clinical case, the authors conducted a non-systematic review of medical literature.
Data synthesisThe results suggest that the following data should be used as a warning for early diagnosis of affected children: (a) combined urinary tract abnormalities (chromosomal abnormalities; sequence of malformations [VACTERLand Prune-Belly]; and musculoskeletal, digestive tract, heart, and nervous system malformations); (b) previous history (congenital anomalies of the kidney and urinary tract [CAKUT] in the family, low birth weight, and oligoamnios); (c) clinical signs (polyuria/nocturia, urinary tract infection, systemic arterial hypertension, failure to thrive, weak urinary stream, difficulty to start urination, distended bladder, non-monosymptomatic enuresis, urinary/urge incontinence, and bowel and bladder dysfunction); and (d) pre- and postnatal ultrasonographic alterations (increased anteroposterior diameter of the renal pelvis, mainly in the third trimester of pregnancy; single kidney; hydronephrosis associated with other abnormalities; and hydronephrosis with parenchymal involvement in the post-neonatal assessment).
ConclusionThe suggestions shown here can help the pediatrician to establish clinical hypotheses for the early diagnosis of developmental abnormalities of the genitourinary tract without resorting to expensive and invasive procedures.
As anormalidades do desenvolvimento do trato geniturinário são a principal causa de doença renal crônica (DRC) em crianças. O diagnóstico dessa doença no Brasil é formulado de maneira incompleta e tardia, o que resulta em aumento na morbi-mortalidade nessa faixa etária. O diagnóstico precoce dessa condição é prerrogativa dos pediatras generalistas e o objetivo desse trabalho foi revisar os sinais e sintomas clínicos associados às anormalidades do desenvolvimento do trato geniturinário.
Fontes dos dadosA partir da descrição de um caso clínico simbólico, realizamos uma revisão não sistemática da literatura médica.
Síntese dos dadosOs resultados sugerem que os seguintes dados devem ser utilizados como alerta para o diagnóstico precoce das crianças acometidas: a) anomalias do trato urinário compostas (anomalias cromossômicas, sequências de malformações – VACTERL e Prune-Belly, malformações musculoesqueléticas, do trato digestivo, cardíacas e do sistema nervoso), b) antecedentes (anomalias congênitas do rim e trato urinário (CAKUT) na família,baixo peso ao nascer e oligoâmnio), c) sinais clínicos (polaciúria/noctúria, infecção urinária, hipertensão arterial sistêmica, baixo ganho de peso, jato urinário fraco, dificuldade para iniciar a micção, bexigoma, enurese não monossintomática, urge/incontinência urinária, disfunção do intestino e da bexiga)e d) alterações ultrassonográficas ante e pós-natais (diâmetro ântero-posterior da pélvis renal aumentado principalmente no terceiro trimestre da gestação, rim único, hidronefrose associada a outras anomalias e hidronefrose com comprometimento de parênquima na avaliação pós-neonatal).
ConclusãoAs sugestões apresentadas podem ajudar o pediatra a estabelecer hipóteses clínicas para o diagnóstico precoce das anormalidades do desenvolvimento do trato geniturinário sem utilização de metodologias caras e invasivas.
The patient, H.O.R.S., aged 12 years and 3 months, was born at term with no complications during childbirth. He was discharged 72h after delivery in the mother's company, who was instructed to carry on the follow-up with a specialist, as the prenatal ultrasonography had shown the presence of bilateral hydronephrosis with anteroposterior renal pelvis diameter of approximately 12mm. However, after birth, the parents were lost to follow-up with the nephrologist.
The consultations with the pediatrician were irregularly attended. At 8 years old, the child was referred for consultation with a hematologist and an endocrinologist due to stunting and anemia unresponsive to treatment with oral iron; these specialists did not find the cause for the alterations.
Only at the age of 10 years and 11 months was the child diagnosed with chronic kidney disease (CKD) secondary to bilateral ureteropelvic junction stenosis. At the time of diagnosis, he had already shown signs of chronic nephropathy with bilateral tapering of the renal parenchyma at the ultrasonographic assessment. The parents stated that at the age of 2 years the child already had daytime sphincter control, but they observed polyuria, nocturia, and excessive water consumption. There were no alterations in urinary stream or episodes of urinary tract infection.
At 11 years and 3 months, he underwent a pyeloplasty with double J catheter implantation on the right in order to prolong the conservative treatment and delay the onset of renal replacement therapy.
At the age of 12 years and 3 months, one year and four months after the diagnosis, he underwent preemptive kidney transplantation from a living donor (his mother).
DiscussionThe case described above is factual and common, evidencing the disturbing problem of late diagnosis of CKD in children in Brazil. CKD is defined as the existence of any kidney injury, associated with varying degrees of glomerular filtration rate decrease, as shown in Table 1.1 Stage 5 CKD, requiring renal replacement therapy (RRT; whether dialysis or transplantation), is a public health problem and there is evidence of the increasing incidence and prevalence of this condition.2 In 2010, the number of individuals receiving RRT worldwide was estimated at 2.7 million, but it is also estimated that the similar number of patients who had the indication for RRT did not receive it due to lack of access to the treatment, especially for financial reasons.3 It is further estimated that the proportion of CKD patients in stages 2 to 4 is much higher than that of CKD patients in stage 5.4
Classification of chronic kidney disease stages.
| Stage | Description | GFR (mL/min/1.73m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | >90 |
| 2 | Kidney damage with mild decrease in GFR | 60–89 |
| 3 | Kidney damage with moderate decrease in GFR | 30–59 |
| 4 | Kidney damage with severe decrease in GFR | 15–29 |
| 5 | Renal failure | <15 |
Fortunately, the proportion of children with CKD is much lower than that of adults, but CKD in children has devastating consequences on growth, weight, height, and intellectual development, resulting in increased morbidity and mortality.4 The prevalence of pediatric CKD in Brazil is lower than that reported in developed Western countries, probably due to the lack of diagnosis. A study carried out in Brazil in 2012 estimated a total of approximately 1300 pediatric patients on dialysis in the country, resulting in a prevalence of 20 cases per million age-related population (pmarp).5 In Europe, for instance, records show a prevalence rate of 65 pmarp, whereas in the United States, these numbers reach 85 pmarp.6
In developed countries, the main cause of CKD in childhood are inherited diseases, particularly congenital anomalies of the kidneys and urinary tract (CAKUT), which amount to approximately 30% to 60% of children with CKD on RRT.4,6 In contrast, the main etiological diagnosis that was given to children on dialysis in Brazil was “unknown cause”, comprising 32% of the cases, which is much higher than that reported in Western countries, suggesting that this condition is underdiagnosed in Brazil.5 Another noteworthy aspect is the time between CKD diagnosis and the start of RRT, which in Brazil was 14 months in the Southeast, nine months in the Midwest, and zero months in the Northeast, suggesting a delay in establishing the diagnosis, which also impairs the final prognosis of these children.5
In the aforementioned clinical case, the patient already had anemia and stunting at the time of the diagnosis, which are signs of the late diagnosis. This deserves discussion because, similarly to this case, many others can go undiagnosed, causing harm to the patients. The early diagnosis of CKD is an opportunity for the generalist pediatricians, not for the specialists. It allows for the initiation of medical therapies and indication of surgical procedures when necessary, which can slow the progression of CKD and the need to start RRT.
The genetic diagnosis of CAKUT is challenging and not yet accessible, due to the high costs of genetic tests and the complexity of interactions among multiple candidate genes to explain the diseases,7 and will not be discussed here. This study aimed to review the clinical signs and symptoms associated with the most prevalent CAKUT in childhood, in order to provide the generalist pediatrician with elements for the early diagnosis and treatment, aiming to attain a better prognosis of affected children.
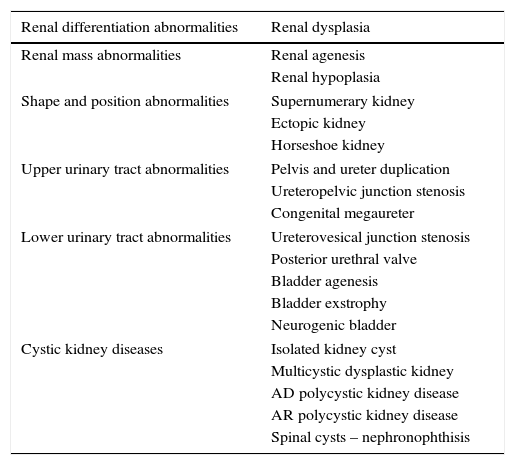
Classification of congenital abnormalities of the kidneys and urinary tractThe CAKUT are a heterogeneous group of diseases of the kidney and urinary tract development (Table 2). In a German study of 30,940 newborns, stillbirths, and miscarriages/abortions, it was estimated that approximately 7% had major malformations. CAKUT were the second most frequent abnormality, with an estimated prevalence of 1.6% of the total.8 In a Brazilian study of 6245 perinatal and pediatric autopsies, the authors observed that CAKUT were present in 2.9% of the cases.9
Congenital abnormalities of the kidneys and urinary tract.
| Renal differentiation abnormalities | Renal dysplasia |
|---|---|
| Renal mass abnormalities | Renal agenesis |
| Renal hypoplasia | |
| Shape and position abnormalities | Supernumerary kidney |
| Ectopic kidney | |
| Horseshoe kidney | |
| Upper urinary tract abnormalities | Pelvis and ureter duplication |
| Ureteropelvic junction stenosis | |
| Congenital megaureter | |
| Lower urinary tract abnormalities | Ureterovesical junction stenosis |
| Posterior urethral valve | |
| Bladder agenesis | |
| Bladder exstrophy | |
| Neurogenic bladder | |
| Cystic kidney diseases | Isolated kidney cyst |
| Multicystic dysplastic kidney | |
| AD polycystic kidney disease | |
| AR polycystic kidney disease | |
| Spinal cysts – nephronophthisis | |
AD, autosomal dominant; AR, autosomal recessive.
In addition to its role as a main cause of CKD in children, it is understood that CAKUT represent an important CKD etiology also in adults. A recent study of 212,930 adult individuals with CKD showed that patients with CAKUT started RRT at 31 years old, whereas individuals with other etiologies required RRT only at 61 years of age.10 This suggests that adults with kidney failure have CAKUT, which, as during childhood, can be identified and treated early, increasing the importance of this diagnosis.
Subsidies for the diagnosis of congenital abnormalities of the kidneys and urinary tractAssociated malformationsCAKUT have very different genetic causes and factors, such as hereditary chromosomal abnormalities and mutations in genes that regulate the development of the urinary tract, which can be the underlying cause of the malformation. Moreover, environmental effects on fetal development are also important, resulting in a complex association between genotype and phenotype in these diseases. As a starting point to try characterizing this complexity, it is important to consider that CAKUT may appear alone or may be associated with other malformations or syndromes.
In a recent study with 346,841 abortions/miscarriages, stillbirths, or live births, it was estimated that 34% of the 1678 observed cases of CAKUT had other non-urinary associated malformations. The distribution of these associations is shown in Table 3,11 and their importance for the generalist pediatrician is that their presence should be a warning to search for CAKUT cases.
Malformations associated with CAKUT found in 346,381 cases in France from 1979 to 2004 (total of 1678 CAKUT, of which 563 had other associated malformations).
| Chromosomal abnormalities (n=119) | Trisomy 18 (n=33) |
| Trisomy 21 (n=26) | |
| Trisomy 13 (n=24) | |
| Turner syndrome (n=11) | |
| Others (n=25) | |
| Non-chromosomal associations (n=168) | VACTERL (n=56) |
| Meckel-Gruber (n=36) | |
| Prune-Belly (n=23) | |
| Others (n=53) | |
| Unknown associations (n=276) | |
| Skeletal muscle tissue (n=97) | Polydactyly/syndactyly (n=45) |
| Shortening of limbs (n=30) | |
| Others (n=22) | |
| Digestive tract (n=91) | Anal atresia (n=38) |
| Pathological rotation (n=29) | |
| Others (n=24) | |
| Cardiac malformations (n=88) | VSD (n=42) |
| ASD (n=21) | |
| Others (n=25) | |
| Nervous system (n=60) | Spina bifida (n=21) |
| Encephalocele (n=15) | |
| Microcephaly (n=8) | |
| Others (n=16) | |
| Head and neck (n=53) | Facial dysmorphia (n=21) |
| Hypertelorism (n=19) | |
| Others (n=13) | |
| Respiratory system (n=28) | Pulmonary hypoplasia (n=21) |
| Others (n=7) | |
| Abdominal wall (n=23) | Omphalocele (n=14) |
| Gastroschisis (n=9) | |
| Others (n=29) | Cleft lip (n=23) |
| Diaphragmatic hernia (n=6) | |
VSD, ventricular septal defect; ASD, atrial septal defect.
Modified from Stoll et al.11
Past medical history in the antenatal period that is associated with CAKUT most often includes oligoamnios, prematurity, low birth weight, and intrauterine growth restriction. A study carried out in Japan showed an association between perinatal history and CAKUT, suggesting signs to indicate the screening of children. Of the assessed data (14% of patients with CAKUT were preterm, 18% had low birth weight, 79% showed failure to thrive, 18% had asphyxia, 8% had oligoamnios, and 12% had jaundice), it was found a total of 82% with a history of oligoamnios and failure to thrive, suggesting these two signs are markers to select individuals at higher risk for CAKUT, with a specificity of 95%.12 In a Brazilian study that assessed previous historical data, the latter were a predictor of mortality associated with CAKUT in 29,653 newborns between 1996 and 2006. This study showed 524 cases of CAKUT, corresponding to 17.7 per 1000 live births and the risk factors for mortality were oligoamnios, CAKUT associated with other malformations, low birth weight, and prematurity.13 Another noteworthy past medical history is CAKUT in the family. A recent study showed that 14% of 107 cases of CAKUT had a family history with a confirmed diagnosis in at least one family member.14
In a study involving 1994 patients with CKD (diagnosed before the age of 21) and 20,032 controls, low birth weight and maternal pre-gestational diabetes were significantly associated with the risk of renal dysplasia/aplasia.15
The literature is quite scarce on the description of warning signs and symptoms of urinary system impairment. In the case of CAKUT, the clinical expression of malformations can conceptually be due to: (a) impairment of the upper urinary system leading to renal dysfunction, and can be expressed as polyuria, pollakiuria, nocturia, urinary tract infection, systemic arterial hypertension, and low weight gain14,16–18 or (b) signs of lower urinary tract impairment with urinary tract obstruction, such as weak urinary stream, difficulty to start urination, distended bladder, non-monosymptomatic enuresis, urinary urge/incontinence, and bowel and bladder dysfunction.19
The controversy on the use of episodes of urinary tract infection as a CAKUT indicator is quite intense, but it appears that some situations represent a higher probability of association with CAKUT. Age younger than 2 years and high fever (higher than 38.3°C) are the factors that can predict urological abnormalities.20
Finally, another evidence that is relevant to allow for the early diagnosis of congenital nephropathy is blood pressure. It is known that hypertension is more common in children with nephropathy and, conversely, that blood pressure control has an effective therapeutic response by slowing CKD progression in children.21 These associations reinforce the importance of the recommendation of blood pressure measurement in children, which should be performed in all pediatric visits, from the age of 3 years onwards, and in children younger than 3 years in the presence of any of the following situations: (a) preterm birth, low birth weight or neonatal complications requiring admission to the neonatal intensive care unit; (b) congenital heart diseases; (c) urinary tract infection, hematuria, or proteinuria; (d) known kidney disease or urologic malformations; (e) family history of congenital kidney disease; (f) solid organ transplantation; (g) malignancies or bone marrow transplantation; (h) systemic diseases associated with hypertension; and (i) evidence of high blood pressure.22
Antenatal ultrasonographic alterationsThe use of ultrasonography in the monitoring of pregnancy and neonates began in the late 1970s. Currently, gestational ultrasonography is part of the medical routine in most countries, with at least two examinations during pregnancy to monitor fetal development and identify possible malformations.23 Gestational ultrasonography greatly improved the diagnosis and prognosis of children with CAKUT, reaching a level of sensitivity for the detection of obstructive uropathies between 80% and 90%. In a European study conducted in 12 countries, the sensitivity to renal malformation detection was 81.2%.23
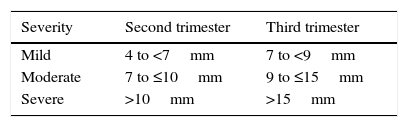
Hydronephrosis is the ultrasonographic finding most often identified and described. Antenatal hydronephrosis is the presence of one or both kidneys with some degree of pyelocalyceal system dilation in the fetus. The diagnosis is attained through obstetric ultrasonography, using as the main parameter the anteroposterior diameter (APD) of the renal pelvis and the presence of caliectasis. There is hydronephrosis when the pelvis APD is above certain limits, but there is no consensus on the definition of these limits yet. The most frequently cited values, above which there is a greater risk of renal function deterioration or need for surgery, are those proposed by the Society for Fetal Urology (SFU),24 which are summarized in Table 4.
The highest positive predictive value (PPV) is found in the examination performed in the third trimester of pregnancy. The PPV for APD values >7mm in the third trimester is 69% vs. 49% for APD>4mm in the second trimester.4 The reported incidence of fetal disease varies from 1% to 5% of pregnancies, depending on diagnostic criteria; it is more frequent in boys (3 to 4/1) predominantly unilateral, and only 20% of cases will have postnatal clinical significance.
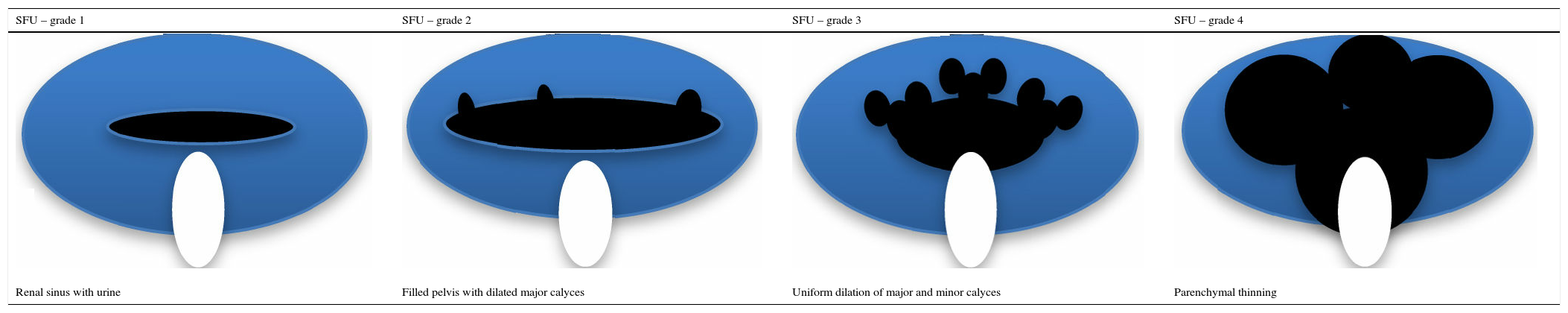
Another ultrasonographic finding that is important for the diagnosis and prognosis of children with CAKUT is the hydronephrosis classification proposed by the SFU, which is based on the degree of pyelocalyceal dilation and also takes parenchyma integrity into account. Grade 0 represents a normal ultrasonography; the other grades are shown in Table 5.
Graphic representation and classification of the ultrasonographic alterations in hydronephrosis (adapted from Timberlake & Herndon25).
| SFU – grade 1 | SFU – grade 2 | SFU – grade 3 | SFU – grade 4 |
|---|---|---|---|
| Renal sinus with urine | Filled pelvis with dilated major calyces | Uniform dilation of major and minor calyces | Parenchymal thinning |




SFU, Society of Fetal Urology.
This classification system was developed for ultrasonography performed after birth. However, it has been applied to antenatal ultrasonography, and there is evidence that children with grades 3 and 4 of hydronephrosis have increased risk of need for surgery and renal function deterioration.25
Regardless of the classification of hydronephrosis, the postnatal follow-up of children with images of hydronephrosis detected during the gestational period shows resolution in approximately 50% of cases, caliectasis with no obstructive factor including extrarenal pelvis in 15%, ureteropelvic junction stenosis in 11%, vesicoureteral reflux in 9%, megaureter in 4%, multicystic dysplastic kidney in 2%, ureterocele in 2%, renal cysts in 2%, and posterior urethral valve in 1%.25–27
Other imaging findings obtained at the gestational ultrasonography that help with the diagnosis and prognosis of children with CAKUT and are also associated with renal function deterioration are: (a) the finding of single kidney, in which case 1/3 of the affected individuals has associated CAKUT,28,29 (b) hydronephrosis associated with other anomalies, when the probability of CKD at 2 years of age is 15% vs. only approximately 2% in children with isolated hydronephrosis30 and (c) bilateral hydronephrosis, which represents a risk of renal function deterioration that is 9.4-fold higher than cases with unilateral alteration.31
In addition to the aspect and degree of hydronephrosis, other ultrasonographic findings should be taken into account, such as: (a) ureteral dilatation, which is present in megaureter; ureterovesical-junction stenosis, posterior urethral valve, and absent in the ureteropelvic junction stenosis; (b) kidney size, which can be affected in renal hypo/dysplasia, or even in hydronephrosis; (c) bladder size and bladder wall thickness, with or without formation of trabeculae, alterations that may be present in the hypertrophic or hypotonic bladder; (d) increased post-voiding residual urinary volume, associated with poor bladder function; and (e) ureterocele, usually present in ureteral duplicity.32
ConclusionsThis study aimed to review the medical literature to list the main clinical data that might be useful in pediatric practice for the early identification of patients with CAKUT. This diagnosis is important, since CAKUT are the most common cause of CKD in children. In the present epidemiological scenario, characterized by a decrease in morbidity and mortality secondary to infectious diseases, chronic diseases in general (and particularly CKD) have become more important in clinical practice, which should become even more prominent with time.2
Current evidence shows that CKD has its origins in intrauterine life, and at present it is known that the number of nephrons in each kidney can vary greatly between individuals, and that a lower number of kidney functional units increases the risk of CKD and cardiovascular disease.33,34 This consideration is even more important in children with urinary system development abnormalities, in which nephrogenesis is often disturbed, resulting in a lower number of nephrons. These findings anticipate to the pediatric range the moment of greatest interest to attain an early diagnosis of patients at risk for CKD.
The time to perform this diagnostic search should be as early as possible, preferably before birth. It is known that even in situations where well-organized follow-up services are available, incomplete adherence of families to the necessary procedures for diagnostic research is a frequent phenomenon.35
Some countries seek the early diagnosis of risk for CKD using compulsory programs of urinary sediment tests or imaging assessment at certain ages,36,37 but the cost-effectiveness analyses of such programs never conclusively demonstrated that this initiative was advantageous.38 In a situation such as this one, clinical evaluation is an invaluable resource to screen which patients should be studied through more detailed laboratory tests. This strategy appears to be particularly accurate in countries with limited resources in the health care area. In Brazil, with the exception of the Southeast region, the unknown cause is the most common diagnosis of advanced CKD in children,5 suggesting that there is an important gap to be filled in order to better categorize the diagnosis of CKD. It is plausible that many children reach the final stage of the CKD evolution without having access to diagnosis of CAKUT, which is the most important cause of this disease worldwide.
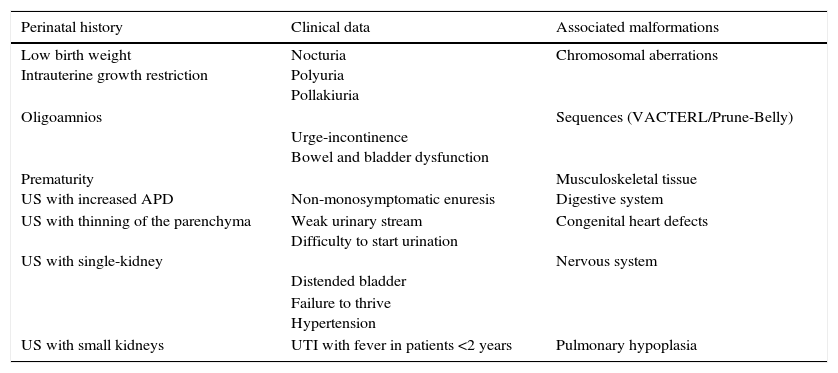
A noteworthy observation in this review was that there are no studies with a robust methodology to provide answers based on solid evidence about the subject. Most of the reviewed studies were descriptive and single-centered and, therefore, the findings summarized herein must be considered as preliminary data. Warning signs for the identification of children with CAKUT are summarized in Table 6.
Warning signs for the diagnosis of CAKUT in children.
| Perinatal history | Clinical data | Associated malformations |
|---|---|---|
| Low birth weight Intrauterine growth restriction | Nocturia Polyuria Pollakiuria | Chromosomal aberrations |
| Oligoamnios | Urge-incontinence Bowel and bladder dysfunction | Sequences (VACTERL/Prune-Belly) |
| Prematurity US with increased APD | Non-monosymptomatic enuresis | Musculoskeletal tissue Digestive system |
| US with thinning of the parenchyma US with single-kidney | Weak urinary stream Difficulty to start urination Distended bladder | Congenital heart defects Nervous system |
| Failure to thrive Hypertension | ||
| US with small kidneys | UTI with fever in patients <2 years | Pulmonary hypoplasia |
US, ultrasonography; APD, anteroposterior diameter; UTI, urinary tract infection.
The authors believe that, even with the methodological limitations of the reviewed studies, the suggestions can help pediatricians to establish clinical hypotheses for the early diagnosis of abnormalities of the genitourinary tract development, by using clinical data and information from the gestational ultrasonography, without resorting to expensive and invasive methods. As there are no prospective data to evaluate which clinical signs and symptoms listed here are more important to identify the risk, the authors believe that, for the moment, the occurrence of any of them should be an indication to investigate children regarding the possible existence of CAKUT, as early as possible.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Nogueira PC, Paz IP. Signs and symptoms of developmental abnormalities of the genitourinary tract. J Pediatr (Rio J). 2016;92(3 Suppl 1):S57–63.





