
To discuss the etiology and growth consequences of small size at birth and the indications, effects, and safety of biosynthetic growth hormone therapy in children born small for gestational age.
Source of dataA comprehensive and non-systematic search was carried out in the PubMed, LILACS, and SciELO databases from 1980 to the present day, using the terms “small for gestational age,” “intrauterine growth restriction,” and “growth hormone”. The publications were critically selected by the authors.
Data synthesisAlthough the majority of children born small for gestational age show spontaneous catch-up growth during the first two years of life, some of them remain with short stature during childhood, with high risk of short stature in adult life. Treatment with growth hormone might be indicated, preferably after 2–4 years of age, in those small for gestational age children who remain short, without catch-up growth. Treatment aims to increase growth velocity and to reach a normal height during childhood and an adult height within target height. Response to growth hormone treatment is variable, with better growth response during the pre-pubertal period.
ConclusionsTreatment with growth hormone in short children born small for gestational age is safe and effective to improve adult height. Efforts should be done to identify the etiology of small size at birth before treatment.
Discutir a etiologia e as consequências para o crescimento e as indicações, os efeitos e segurança da terapia com hormônio de crescimento biossintético em crianças pequenas para idade gestacional.
Fonte dos dadosUma busca abrangente e não sistemática foi feita nas bases de dados PubMed, LILACS e SciELO de 1980 até a presente data, com os termos “small for gestational age” (pequeno para a idade gestacional), “intrauterine growth restriction” (restrição de crescimento intrauterino) e “growth hormone” (hormônio do crescimento). As publicações foram selecionadas criticamente pelos autores.
Síntese dos dadosEmbora a maioria das crianças nascidas pequenas para idade gestacional apresente recuperação espontânea do crescimento durante os dois primeiros anos de vida, algumas delas permanecem com baixa estatura durante a infância, com alto risco de baixa estatura na vida adulta. O tratamento com hormônio de crescimento pode ser indicado, preferencialmente após os dois aos quatro anos, naquelas crianças sem recuperação espontânea do crescimento e com baixa estatura. Seus objetivos são aumentar a velocidade de crescimento e atingir uma altura normal durante a infância e uma altura adulta dentro da altura-alvo. A resposta ao tratamento com hormônio de crescimento é variável, com melhor resultado se iniciado durante o período pré-puberal.
ConclusõesO tratamento com hormônio de crescimento em crianças baixas nascidas pequenas para idade gestacional é seguro e eficaz para melhorar a estatura adulta. Esforços devem ser feitos para identificar a etiologia do nascimento pequenas para idade gestacional antes do tratamento.
About 20 million children are born worldwide with low birthweight (<2500g) every year, but this definition encompass children born preterm (<37 weeks of gestation) and children born small for gestational age (SGA).1 The estimated prevalence of children born at term and SGA varies in several countries and regions, ranging from 2.3% in the United States,2 5.5% in Sweden,3 3.4% in Japan,4 12.5% in Latin America,5 and 44.5% in South Asia.5 Among children born preterm, especially below 34 weeks, the prevalence of SGA is higher.4–6
Multiple criteria have been used to define SGA. In 1995, the World Health Organization (WHO) recommended the definition of SGA as birthweight less than the 10th percentile for gestational age, due to the increased perinatal and neonatal risks of these children compared to newborns with appropriate size.7,8 The consensus of the Pediatric Endocrinology Societies9,10 recommended that SGA should be defined as birth weight and/or birth length equal to or less than −2.0 SD for sex and gestational age, in order to include mainly those with increased risk of growth and metabolic disturbances.9
Besides these criteria, accurate gestational dating and measurements at birth are necessary for the definition of SGA, in addition to the growth international standard chosen. The influence of the standard selected for the definition of SGA is critical. Recently, the INTERGROWTH-21st birth size standards were validated with the possibility to use for children born between 33 and 42 postconceptional weeks.11 Approximately 24% of the newborns considered SGA according to the INTERGROWTH-21st standards were considered appropriate for gestational age (AGA) according to the Fenton preterm growth charts.12 However, the INTERGROWTH-21st is not recommended for gestational ages lower than 33 weeks. In this case, the current recommendation of the Brazilian Society of Pediatrics for preterm growth monitoring is to consider the growth channel achieved after the stabilization of neonatal weight loss, but it do not recommend any specific reference for size at birth definition.13 The intrauterine (fetal) growth charts have not been considered a good option to evaluate size at birth,14 although Fenton & Kim growth Charts15 are still used for this purpose in very or extremely preterm children.
Most children born SGA show spontaneous catch-up to weight and height above −2.0 standard deviation scores (SDS) during the first 2 years of life16,17 and will reach an adult height at the normal range for the population and/or their target height. About 10% of the children born SGA fail to show catch-up growth (CUG) and account for approximately 20% of all cases of short stature during adult life.18 Some of these children may benefit from growth hormone (GH) treatment.
In this review, the authors discuss the etiology and consequences of small size at birth, as well as the effects and safety of GH therapy in children born SGA who did not present CUG spontaneously.
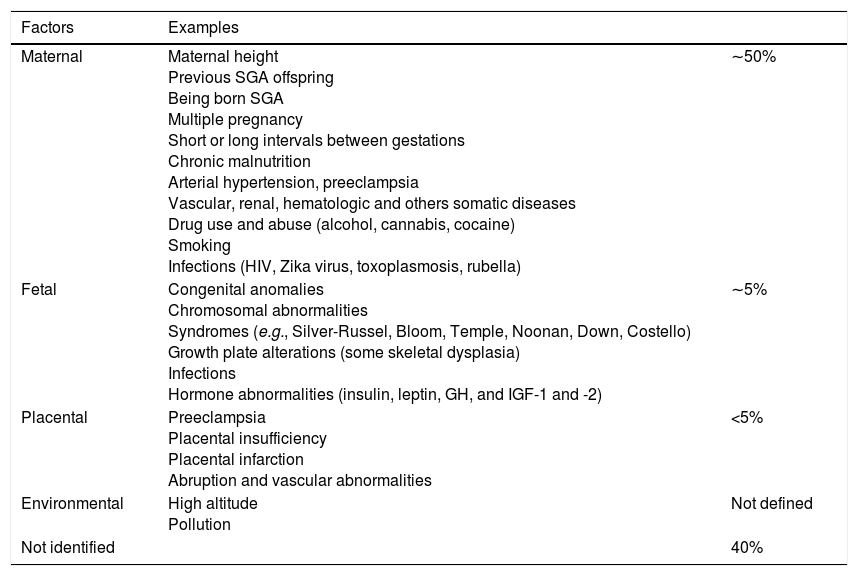
Etiology and classificationSmall size at birth has multifactorial etiology, including fetal, placental, maternal, and environmental factors. In about 40% of the cases, the reason of small size at birth is not identified (Table 1). Many of these situations might induce poor fetal growth with intrauterine growth retardation (IUGR). However, IUGR should not be used as synonyms of SGA: SGA refers to body size at birth and the IUGR refers to the reduction of the growth velocity documented by at least two fetal measurements.9
Etiology of small for gestational age birth.
| Factors | Examples | |
|---|---|---|
| Maternal | Maternal height Previous SGA offspring Being born SGA Multiple pregnancy Short or long intervals between gestations Chronic malnutrition Arterial hypertension, preeclampsia Vascular, renal, hematologic and others somatic diseases Drug use and abuse (alcohol, cannabis, cocaine) Smoking Infections (HIV, Zika virus, toxoplasmosis, rubella) | ∼50% |
| Fetal | Congenital anomalies Chromosomal abnormalities Syndromes (e.g., Silver-Russel, Bloom, Temple, Noonan, Down, Costello) Growth plate alterations (some skeletal dysplasia) Infections Hormone abnormalities (insulin, leptin, GH, and IGF-1 and -2) | ∼5% |
| Placental | Preeclampsia Placental insufficiency Placental infarction Abruption and vascular abnormalities | <5% |
| Environmental | High altitude Pollution | Not defined |
| Not identified | 40% |
SGA, small for gestational age; HIV, human immunodeficiency virus; GH, growth hormone; IGF, insulin-like growth factor.
Children born SGA can be sub-classified into SGA due to weight (SGA-w), to length (SGA-l), or to both (SGA-w/l), all with different growth prognosis.9 For example, the relative risk of short stature during adult life is 7.1 times higher for those born SGA-l and 5.2 for those born SGA-w, when compared with children born AGA.19 According to the period of pregnancy in which the fetal growth impairment occurred, SGA children can also be sub-divided into symmetrical, when it occurred at the beginning of gestation, and asymmetrical, when it occurred mainly during the third trimester. Symmetrical SGA children have proportionally decreased weight, length, and head circumference, and asymmetrical SGA have low weight but normal or near-normal length and head circumference.9
Among the abnormalities resulting in small size at birth cited in Table 1, some deserve more attention when discussing GH treatment. Silver-Russell (SRS) and Temple syndrome are imprinting disorders that are associated with fetal and postnatal growth retardation. SRS has a global incidence of 1:30,000 to 1:100,000 births and is characterized by triangular shaped head with frontal bossing, clinodactyly, asymmetry of face and/or body, fetal and postnatal growth retardation, and feeding difficulties.20 Almost all are born SGA, remain short during childhood, and reach a mean adult height around −4.0 SDS. Children with Temple syndrome present obesity, hypotonia, early puberty, and short stature.21 Maternal uniparental disomy (UPD) of chromosome 20 causes a syndrome characterized by fetal and postnatal growth retardation and feeding difficulties that differ from SRS because there is not asymmetry and relative macrocephaly.22 Bloom syndrome is a breakage chromosomal syndrome with severe fetal and postnatal growth retardation, an erythematous facial rash after sun exposure, microcephaly and malar hypoplasia, immunodeficiency, and an increased risk of cancer at an early age.23 Fanconi anemia, neurofibromatosis type 1, and ataxia-telangiectasia are also conditions with short stature and increased risk of malignancies.23 In addition, some genetic defects affecting growth plate might be associated with small size at birth and short stature, mainly in those born SGA-l, for instance heterozygous mutations of ACAN (encoding Agrecan) and IHH (Indian hedgehog) genes, resulting in skeletal dysplasia. Nevertheless, SGA is an uncommon finding in achondroplasia.22 In summary, extensive diagnostic work-up and identification of the cause of small size at birth is mandatory before the decision of GH treatment in short children born SGA in order to increase efficacy and ensure treatment safety.
Postnatal growthCUG is defined as a growth rate greater than the median for chronological age and gender for reaching a height above −2.0 SD or 3rd percentile, but target height should be also considered.24 Most children born SGA show spontaneous CUG to weight and height above -2.0 SD during the first 2 years of life,16,17 the majority during the first 2–3 months,19,25 reaching 86% until 9–12 months of life for SGA with birth weight <−2 SD and 79% when the 5th percentile is used.26 When children born SGA and preterm were evaluated, 82.5% showed CUG until 2 years of age, with later CUG for those born with lower birth length SDS. Birth length and birth weight SDS had a significant positive association with CUG at 2 years in full-term and preterm SGA children, with no association with gestational age in those born preterm.16
About 10%–15% of children born SGA remain short, and the reason for the insufficient CUG might be related to genetic abnormalities, disturbances of GH secretion, and/or reduced insulin-like growth factor 1 (IGF-1) levels,17,27 although most of the SGA children are not GH-deficient. The lack of spontaneous catch-up in length within the first four months of life can be used to early identify those SGA children with higher risk of short stature at 5 years of age.28
Although approximately 10% of adults born SGA remain with short stature, in only 3.7% stature is below −2.5 SD.29 For those with spontaneous CUG, a mean adult height of −0.7 SD has been reported, whereas for those who remain short throughout childhood, the final height reported was −1.7 SD, 7.5cm less than target height in men and 9.6cm less in women.3 Leger et al. found a deficit in adult height adjusted for target height of 0.8 SD (−3.9cm) in males and 0.9 SD (−3.64cm) in females born SGA, when compared with subjects born AGA (p<0.0001).30 CUG in early childhood and adult height were associated with birth length and parental height.18,29,31 The risk of short stature during adult life was 3.2-fold higher for each decrease of 1 SD in maternal height, 2.2-fold higher for each decrease of 1 SD in paternal height, and 1.3-fold higher for each decrease of 1cm in birth length.29
Precocious or fast puberty has been associated with loss of height potential.32 Precocious adrenarche and precocious pubarche, earlier onset of pubertal development, and earlier peak height velocity were reported in children born SGA,20,33–35 although no difference in age of pubertal onset and growth spurt compared with children born AGA has been reported. Normal age range, earlier and no differences in age of menarche30,33–35 between girls born SGA and those born AGA36,37 were reported. Height at onset of puberty was reported to be less than expected in some studies,30,38 which also reported an accelerated epiphyseal fusion.39–41 Faster pubertal progression was described in girls with a decreased pubertal growth and a deficit in height of approximately 4cm.42
Postnatal interventionNutritional management in infants born SGAThe ideal postnatal growth velocity and weight gain in children born SGA or in preterm infants are not fully established. The expected CUG should be gradual; it is beneficial for neurodevelopment and cognitive functions, but can also be deleterious to metabolism.43 SGA newborns frequently receive nutritional requirements similar to that offer to preterm infants.44 Despite gestational age or size at birth, breastmilk is the best nutrient to newborns and exclusive breastfeeding is recommended for the first 6 months of life.45 Breastfeeding must be promoted to mothers of SGA offspring46,47 and it is a way to prevent chronic diseases and obesity later in life.47
The use of high energy formula for SGA infants has been suggested, as it would improve weight gain and increase head circumference without deleterious effect on linear growth.48 However, rapid weight gain during the first two years of life has been associated with obesity and abnormal body composition later in life, with more visceral and abdominal fat and less lean body mass in children and adults born SGA when compared with AGA subjects.49,50
Growth hormone treatment in short children born SGAGH treatment has been indicated for children born SGA who failed to present spontaneous CUG until 2–4 years of age. The goal of GH treatment is to increase growth velocity and to reach a normal height during childhood and an adult height within target height. Efforts should be made in order to identify the cause of small size at birth. Routine pediatric evaluation is needed, and the necessity of a karyotype and genetic tests should be considered. GH stimulating tests are not required before treatment initiation, except if GH deficiency (GHD) is suspected. During these tests, GH secretion does not accurately predict the response to GH treatment.9 However, children with GHD born SGA had lower growth velocity during the first year of GH treatment when compared with children with GHD born AGA.51
The decision of treatment based on bone age and prediction of adult height should be made with caution in children born SGA. Bone age frequently is less than chronological age during prepubertal years; however, an accelerated rhythm of bone age maturation usually occurs from 6 to 8 years of life9,20 with loss of height potential. Consequently, adult height predictions are frequently overestimated during prepubertal years in short children born SGA.9
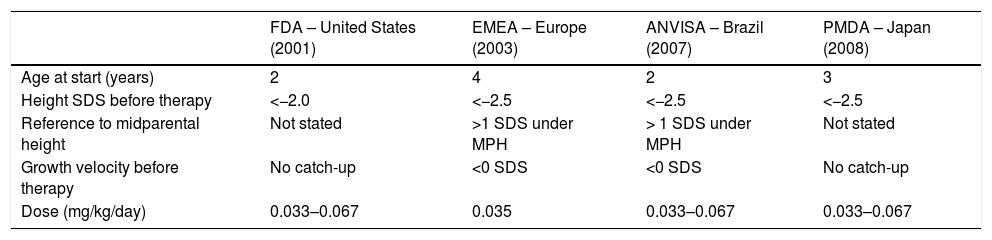
The indication and the dose of GH vary between countries and regions (Table 2). In Brazil, the National Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]) approved GH treatment for children born SGA without CUG in 2007. Children with Silver-Russel syndrome comprise an especial group of children born SGA and might benefit from therapy. In conditions associated with severe hypoglycemia treatment might be initiated before 2 years of age.20 An increment in height velocity should be expected within the first year of therapy with a slight decrease in height velocity during the following years. Treatment is recommended until near final height, when growth velocity falls below 2cm/year and/or bone age is greater than 14 years in girls or 16 years in boys, corresponding to the closure of epiphyseal growth cartilages.9 When growth response is not as expected, a reassessment is indicated including diagnosis, GH dose, compliance evaluation, and the use of drugs that interfere with growth response, such as methylphenidate.52
Indications for GH treatment in short children born small for gestational age.
| FDA – United States (2001) | EMEA – Europe (2003) | ANVISA – Brazil (2007) | PMDA – Japan (2008) | |
|---|---|---|---|---|
| Age at start (years) | 2 | 4 | 2 | 3 |
| Height SDS before therapy | <−2.0 | <−2.5 | <−2.5 | <−2.5 |
| Reference to midparental height | Not stated | >1 SDS under MPH | > 1 SDS under MPH | Not stated |
| Growth velocity before therapy | No catch-up | <0 SDS | <0 SDS | No catch-up |
| Dose (mg/kg/day) | 0.033–0.067 | 0.035 | 0.033–0.067 | 0.033–0.067 |
FDA, Food and Drug Administration; EMEA, European Agency for the Evaluation of Medical Products; ANVISA, Agência Nacional de Vigilância Sanitária; PMDA, Pharmaceuticals and Medical Devices Agency; SDS, standard deviation score, MPH, midparental height.
Although the recommendation is to start GH treatment between 2 and 4 years of age, many short children born SGA start treatment near or at the beginning of puberty. Still controversial and not considered a routine option for short children born SGA,53 the combination of GH with gonadotropin-releasing hormone analogs (GnRHa) might delay epiphyseal maturation and prolong time for GH treatment; moreover, in short children with normal pubertal time, adult height might not be improved.54–56 Lem et al.57 and van der Steen et al.58 showed that the combined therapy in children with stature below 140cm at the beginning of puberty and prediction of adult height <−2.5 SDS resulted in adult height similar to subjects whose puberty started with height above 140cm treated only with GH.57,58 In the Dutch study, height gain during GnRHa+GH therapy was more expressive in boys receiving higher GH doses (0.067 vs. 0.033mg/kg/day; 12.7 vs. 15.2cm, p=0.015), though height gain after GnRHa cessation to adult height was similar with both doses.57 Another study showed that height achieved before puberty onset had a stronger association with pubertal height gain than GH dose, reinforcing the need to start GH treatment and normalize height before puberty.59
The response to GH treatment is variable. Growth response during the first year of therapy is mainly associated with younger age at start of treatment and GH dose. Other determinants of growth response are higher weight SDS at the start of treatment and higher midparental height SDS. For the second and third year, growth velocity during the first year, lower age, higher GH dose, and higher midparental height SDS showed a positive correlation with growth response.60–62
In a systematic review, Maiorana and Cianfarani found that final height of adults born SGA treated with GH was significantly higher than that of the untreated individuals, exceeding by 0.9 SDS, with a mean height gain of 1.5 SDS.63 Similar results were showed by Ross et al.64 and Tanaka et al.41 Less intense gains in adult height have been reported in individuals with SRS compared with non-syndromic subjects born SGA (−2.17 vs. −1.65 SDS, p=0.002),65 and few reports have shown no beneficial effect of GH treatment in adult height.66
Long-term GH therapy for SGA children is safe and well tolerated.41,43 It does not appear to affect time of pubertal onset and progression40; however, a shorter height at onset of puberty40,41 and a less intense pubertal height gain compared to AGA pairs were reported.40,54 High dose of GH was shown to induce acceleration of bone age and onset of puberty in children with idiopathic short stature (ISS).67 Considering the abnormal bone maturation rhythm in children born SGA and possibility of bone acceleration during GH treatment, a closer follow-up of height gain and pubertal progression is recommended. Although GH treatment is not associated with increased risk of malignancy,68 a study showed significantly increase in the incidence of bone and bladder cancers in GH-treated patients without previous malignances, and it was unrelated to cumulative dose or duration of treatment.69 Small size at birth might be associated with syndromes with high risk of malignancy, including Noonan syndrome, neurofibromatosis type 1, Fanconi's anemia, and Bloom syndrome23,43; therefore, greater awareness and prompt assessment of possible cancer symptoms are necessary.70 Extensive and careful diagnostic work-up is necessary to identify these conditions before deciding on GH treatment.
ConclusionTreatment with GH in short children born SGA is safe and effective to improve adult height, especially if initiated before puberty. Combined therapy with GnRHa can be considered for those on puberty and with short stature. Efforts should be made to identify the cause of small size at birth before treatment.
Conflicts of interestBoguszewski MCS has received lecture fees from Pfizer and Sandoz. The other authors declare no conflicts of interest.
Please cite this article as: Cardoso-Demartini AA, Boguszewski MC, Alves CA. Postnatal management of growth failure in children born small for gestational age. J Pediatr (Rio J). 2019;95:S23–S29.




