
The aims of the study were to determine the frequency of hepatobiliary disease in patients with cystic fibrosis and to describe the sociodemographic, clinical, and laboratory profile of these patients.
MethodsThis was a retrospective, descriptive, and analytical study of 55 patients diagnosed with cystic fibrosis, aged between 3 months and 21 years, followed-up from January 2008 to June 2016 in a referral center. Medical records were consulted and sociodemographic, clinical and laboratory data, including hepatobiliary alterations, imaging studies, genetic studies, liver biopsies, and upper digestive endoscopies were registered.
ResultsHepatobiliary disease was diagnosed in 16.4% of the patients and occurred as an initial manifestation of cystic fibrosis in 55.6% of these cases. The diagnosis of hepatopathy occurred before or concomitantly with the diagnosis of cystic fibrosis in 88.9% of the children. All patients with hepatobiliary disease were considered non-white, with a predominance of females (77.8%) and median (IQR) of 54 (27–91) months. Compared with the group without hepatobiliary disease, children with liver disease had a higher frequency of severe mutations identified in the CFTR gene (77.8% vs. 39.6%, p=0.033) and severe pancreatic insufficiency (88.9% vs. 31.6%, p=0.007).
ConclusionThe frequency of hepatobiliary disease was high, with a very early diagnosis of the disease and its complications in the studied series. A statistical association was observed between the occurrence of hepatobiliary disease and the presence of pancreatic insufficiency and severe mutations in the CFTR gene. It is emphasized that cystic fibrosis is an important differential diagnosis of liver diseases in childhood.
Os objetivos do estudo foram determinar a frequência da doença hepatobiliar em pacientes com fibrose cística e descrever o perfil sociodemográfico, clínico e laboratorial destes.
MétodosEstudo retrospectivo, descritivo e analítico de 55 pacientes com diagnóstico de fibrose cística, entre três meses e 21 anos, acompanhados de janeiro de 2008 a junho de 2016 em um centro de referência. Foi realizada consulta aos prontuários médicos, registrando-se os dados sociodemográficos, clínicos e laboratoriais, incluindo-se alterações hepatobiliares, exames de imagem, estudos genéticos, biópsias hepáticas e endoscopias digestivas altas.
ResultadosA doença hepatobiliar foi diagnosticada em 16,4% dos pacientes e ocorreu como manifestação inicial da fibrose cística em 55,6% destes casos. O diagnóstico da hepatopatia ocorreu antes ou concomitante ao diagnóstico da fibrose cística em 88,9% das crianças. Todos os pacientes com doença hepatobiliar foram considerados não brancos, havendo predominância do sexo feminino (77,8%) e mediana (I.I.Q) de idade de 54 (27-91) meses. Em comparação com o grupo sem doença hepatobiliar, as crianças com hepatopatia tiveram maior frequência de mutações graves no gene CFTR identificadas (77,8% vs 39,6%; p=0,033) e de insuficiência pancreática grave (88,9% vs 31,6%; p=0,007).
ConclusãoA frequência de doença hepatobiliar foi elevada, observando-se um diagnóstico muito precoce da mesma e de suas complicações na casuística estudada. Houve associação estatística entre a ocorrência de doença hepatobiliar e a presença de insuficiência pancreática e de mutações graves do gene CFTR. Enfatiza-se que a fibrose cística represente um importante diagnóstico diferencial de hepatopatias na infância.
Cystic fibrosis (CF) is the most common lethal genetic disease in white individuals. Abnormal chlorine and sodium transport through the defective cystic fibrosis transmembrane conductance regulator (CFTR) protein causes an increase in the density of exocrine secretions, affecting several organs.1
The main clinical manifestations of CF are concentrated in the respiratory and gastrointestinal tracts. However, with the increase in the life expectancy of CF patients in the last decades, the prevalence of CF-associated hepatobiliary disease has increased and attracted more attention of patients, family members, and healthcare professionals.2,3
Usually, hepatobiliary manifestations associated with CF start at the end of the patients’ first decade of life, and its prevalence ranges from 5.7% to 39%.4–11 Hepatobiliary disease is the third leading cause of mortality in CF patients, preceded by deaths associated with pulmonary involvement and complications inherent to organ transplants.12 Additionally, some studies have shown that patients with advanced liver disease are at risk of developing and/or exacerbating other extrahepatic CF manifestations, such as malnutrition, diabetes mellitus, hepatic osteodystrophy, and pulmonary disease, leading to increased morbidity caused by the underlying disease.3,13 Thus, hepatobiliary disease has been considered a risk predictor of an unfavorable evolution and prognosis of CF.14
Few studies have been carried out on CF-associated hepatobiliary disease in the Brazilian population, being fundamental to know the disease particularities in Brazil, which has peculiar ethnic/racial characteristics in the different geographic regions. The objectives of the study were to determine the frequency of hepatobiliary disease in CF patients and to describe the sociodemographic, clinical, and laboratory profile of these patients.
MethodsThis was a retrospective, descriptive, and analytical study of patients treated at a referral center in a university hospital. The records of all patients, aged 3 months to 21 years, followed-up between January 2008 and June 2016, with a diagnosis of CF confirmed by two positive sweat tests and/or genetic study, and who had been followed-up for at least three months in this service, were analyzed.
Patients with comorbidities that affected the hepatobiliary system (use of hepatotoxic drugs and/or primary hepatobiliary diseases, or secondary involvement of the liver, gallbladder, and/or spleen) were excluded.
Data were collected from medical records between June and July 2016, consisting of sociodemographic and clinical characteristics – age (months), weight (kg), height (cm), and body mass index (BMI) at CF diagnosis; time of follow-up at the service; presence of hepatobiliary (jaundice, hepatomegaly, splenomegaly, collateral circulation), pulmonary (recurrent pneumonia, wheezing, colonization by Pseudomonas aeruginosa), and gastrointestinal (diarrhea, steatorrhea, low weight gain, meconium ileus) manifestations; Shwachman–Kulczycki score,15 laboratory/imaging tests, and genetic study.
Information on histopathological tests (liver biopsy) and upper digestive endoscopy (UDE) were recorded, when present. Weight, height, and BMI were evaluated by Z-score, according to the World Health Organization (WHO, 2006) classification. Patients with fecal elastase <100μg/g were classified as having severe pancreatic insufficiency. Abdominal ultrasonography (USG) was performed annually in patients older than 7 years, or earlier in those with signs, symptoms, or alterations in laboratory tests suggestive of hepatobiliary disease.
The age at the CF diagnosis was defined as the time of the second positive sweat test or genetic study result. The time of follow-up of each patient corresponded to the period between June 2016 and the first consultation or first hospitalization at the service. Skin color was defined by one of the researchers, medical coordinator of the multidisciplinary CF outpatient clinic, based on the phenotypic characteristics of the patients and their parents.
All patients underwent a genetic study, and the following mutations in the CFTR gene were screened through polymerase chain reaction (PCR) or enzymatic digestion16: F508del, G542X, R1162X, R334W, 3120+1G-A, G551D, and R553X. Infection by cytomegalovirus (CMV) was investigated through serological tests (IgM and IgG antibodies) in patients with hepatobiliary disease criteria, whereas PCR was also performed, when available.
CF-associated hepatobiliary disease was defined as compatible alterations at the liver biopsy or evidence of cholangiopathy (microvesicles, biliary lithiasis, or bile duct stenosis) or the presence, for three consecutive months, of at least two of the following criteria: (1) hepatomegaly on physical examination (liver >2cm from the right costal border, confirmed by abdominal USG); (2) elevation above the upper limit of normal of aspartate aminotransferase and/or alanine aminotransferase and/or gamma-glutamyl transferase levels; and (3) alteration in the abdominal USG (increased hepatic echogenicity, nodularity, irregular liver border, liver fibrosis, or portal hypertension signs).7 The age at diagnosis of hepatobiliary disease was determined at the moment when some of the criteria were observed.
The software SPSS® (IBM SPSS Statistics for Windows, Version 20.0, NY, USA), was used to create the database and carry out the statistical analysis. The descriptive analysis of sociodemographic, clinical, and laboratory characteristics, characterized by measures of central tendency and dispersion (means and standard deviation, median and interquartile range) was performed, as well as the comparison of groups with and without hepatobiliary disease using the Mann–Whitney test for numerical variables and Fisher's exact test for categorical variables. p-values ≤0.05 were considered statistically significant in all analyses.
The study was approved by the Research Ethics Committee of the institution, under registration number 121/2011. All participants and their guardians received verbal and written information about the nature, proposal, and objective of the study and its possible risks and benefits, and signed the informed consent form.
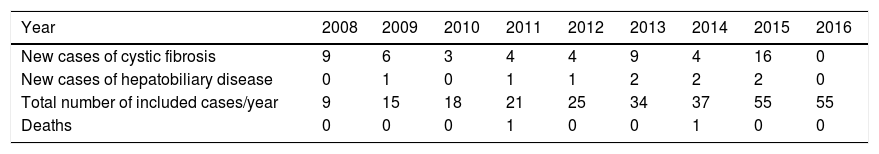
ResultsA total of 55 patients with CF were analyzed. The median (IQR) of the follow-up time of these patients was 35 (15–78) months, ranging from three to 102 months. The distribution of the number of new CF and hepatobiliary disease cases per year of follow-up is shown in Table 1.
Number of new cases of cystic fibrosis and hepatobiliary disease, deaths and total number of cases followed-up between January 2008 and June 2016 in a referral center.
| Year | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 |
|---|---|---|---|---|---|---|---|---|---|
| New cases of cystic fibrosis | 9 | 6 | 3 | 4 | 4 | 9 | 4 | 16 | 0 |
| New cases of hepatobiliary disease | 0 | 1 | 0 | 1 | 1 | 2 | 2 | 2 | 0 |
| Total number of included cases/year | 9 | 15 | 18 | 21 | 25 | 34 | 37 | 55 | 55 |
| Deaths | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
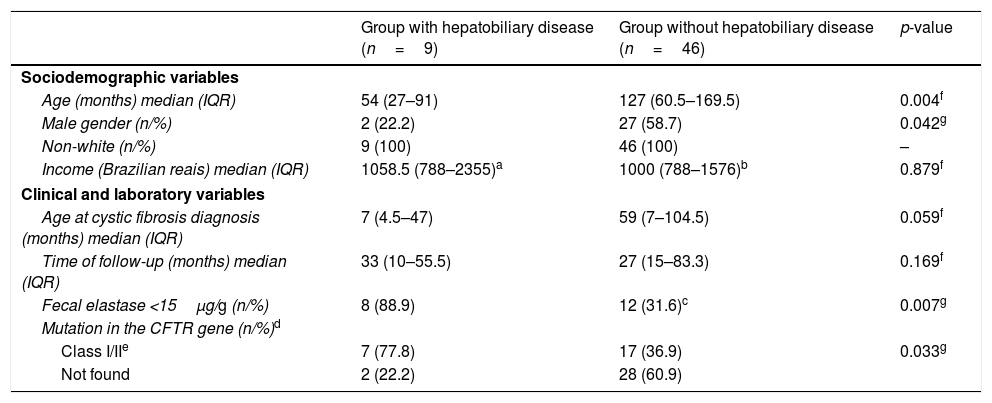
CF-associated hepatobiliary disease was diagnosed in 16.4% (9/55) of CF patients. The median age (IQR) of these patients was 54 (27–91) months, ranging from 10 months to 9 years and 11 months. Seven (77.8%) patients were females; and all were considered non-white. The other sociodemographic, clinical, and laboratory characteristics of the group of CF patients with and without hepatobiliary disease are shown in Table 2.
Sociodemographic, clinical and laboratory characteristics of patients with cystic fibrosis, followed-up between January 2008 and June 2016, in a referral center.
| Group with hepatobiliary disease (n=9) | Group without hepatobiliary disease (n=46) | p-value | |
|---|---|---|---|
| Sociodemographic variables | |||
| Age (months) median (IQR) | 54 (27–91) | 127 (60.5–169.5) | 0.004f |
| Male gender (n/%) | 2 (22.2) | 27 (58.7) | 0.042g |
| Non-white (n/%) | 9 (100) | 46 (100) | – |
| Income (Brazilian reais) median (IQR) | 1058.5 (788–2355)a | 1000 (788–1576)b | 0.879f |
| Clinical and laboratory variables | |||
| Age at cystic fibrosis diagnosis (months) median (IQR) | 7 (4.5–47) | 59 (7–104.5) | 0.059f |
| Time of follow-up (months) median (IQR) | 33 (10–55.5) | 27 (15–83.3) | 0.169f |
| Fecal elastase <15μg/g (n/%) | 8 (88.9) | 12 (31.6)c | 0.007g |
| Mutation in the CFTR gene (n/%)d | |||
| Class I/IIe | 7 (77.8) | 17 (36.9) | 0.033g |
| Not found | 2 (22.2) | 28 (60.9) | |
IQR, interquartile range; N, number of patients.
The median age (IQR) at the diagnosis of hepatobiliary disease associated with CF was 7 months (3–47). This diagnosis occurred before and/or concomitantly with that of CF in 88.9% of the patients, and the presence of hepatobiliary manifestations was among the first signs and symptoms of CF in five of the nine patients (55.6%).
Patients with hepatopathy had a higher frequency of severe mutations in the CFTR gene (77.8% vs. 39.6%, p=0.033) and severe pancreatic insufficiency (88.9% vs. 31.6%, p=0.007) than the group without hepatobiliary disease. Although not statistically significant, the children with hepatobiliary disease had a much earlier CF diagnosis in comparison with the second group, with a median (IQR) of 7 (4.5–47) vs. 59 (7–104.5) months, respectively (p=0.059; Table 2).
Regarding the clinical characteristics of the nine patients with CF-associated hepatobiliary disease, no cases with meconium ileus were observed, and one patient had normal pancreatic function with asymptomatic biliary disease. The others (88.9%) had severe pancreatic insufficiency.
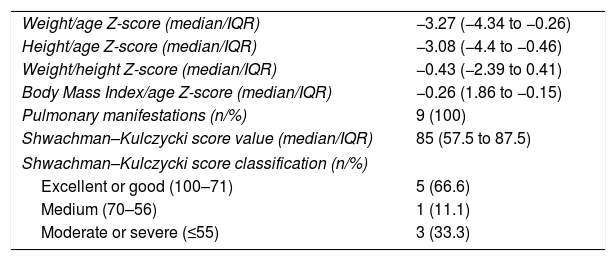
Table 3 shows the clinical characteristics at the time of CF diagnosis of patients with hepatobiliary disease. Some nutritional impairment (Z-score for weight/age and/or height/age and/or BMI/age <−2) was observed in six (66.7%) of these patients. The medians of Z-scores for weight/age and height/age in this group of patients were −3.27 and −3.08, with minimum values of −9.49 and −6.82, respectively. The BMI/age Z-scores were >−2 in 77.8% of the cases, due to the proportional reduction in weight and height values (Table 3).
Clinical characteristics at the time of cystic fibrosis diagnosis of patients with hepatobiliary disease, followed between January 2008 and June 2016, at a referral center.
| Weight/age Z-score (median/IQR) | −3.27 (−4.34 to −0.26) |
| Height/age Z-score (median/IQR) | −3.08 (−4.4 to −0.46) |
| Weight/height Z-score (median/IQR) | −0.43 (−2.39 to 0.41) |
| Body Mass Index/age Z-score (median/IQR) | −0.26 (1.86 to −0.15) |
| Pulmonary manifestations (n/%) | 9 (100) |
| Shwachman–Kulczycki score value (median/IQR) | 85 (57.5 to 87.5) |
| Shwachman–Kulczycki score classification (n/%) | |
| Excellent or good (100–71) | 5 (66.6) |
| Medium (70–56) | 1 (11.1) |
| Moderate or severe (≤55) | 3 (33.3) |
IQR, interquartile range; N, number of patients.
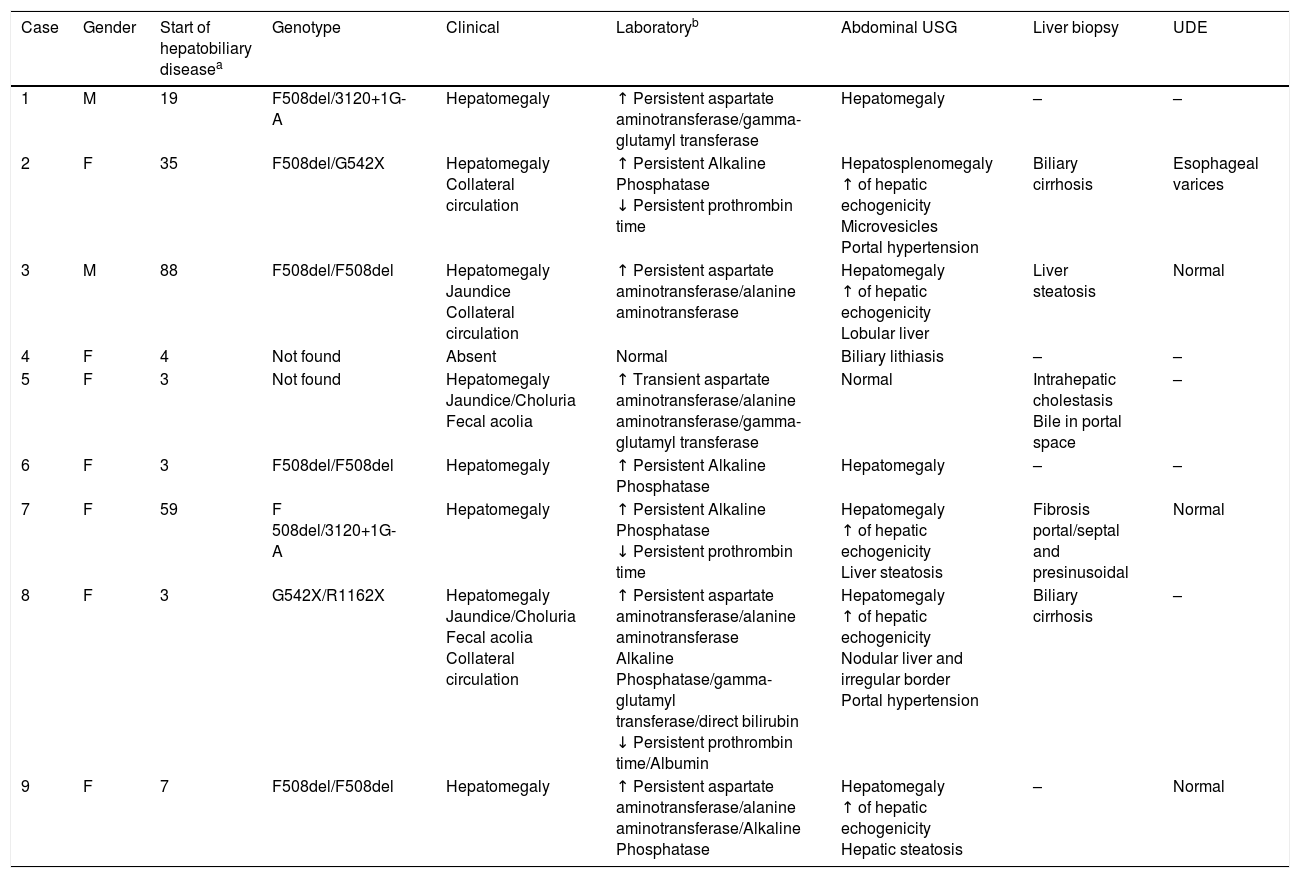
The type of presentation of CF-associated hepatobiliary disease is described in Table 4. Hepatomegaly was the most frequently identified finding, in seven patients (77.8%), followed by increased hepatic echogenicity in five children (55.5%). All patients underwent CMV screening, which was positive in two cases, one of which was also confirmed by PCR. One patient with negative serology had CMV infection confirmed by PCR. These patients had liver biopsy findings compatible with CF-associated hepatobiliary disease. Regarding the complications of hepatopathy, portal hypertension was observed in two patients, splenomegaly in only one case, and esophageal varices in one of the four patients who underwent UDE. Only one child developed signs of acute liver failure.
Clinical presentation of patients from a referral center with hepatobiliary disease associated with cystic fibrosis, between January 2008 and June 2016.
| Case | Gender | Start of hepatobiliary diseasea | Genotype | Clinical | Laboratoryb | Abdominal USG | Liver biopsy | UDE |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 19 | F508del/3120+1G-A | Hepatomegaly | ↑ Persistent aspartate aminotransferase/gamma-glutamyl transferase | Hepatomegaly | – | – |
| 2 | F | 35 | F508del/G542X | Hepatomegaly Collateral circulation | ↑ Persistent Alkaline Phosphatase ↓ Persistent prothrombin time | Hepatosplenomegaly ↑ of hepatic echogenicity Microvesicles Portal hypertension | Biliary cirrhosis | Esophageal varices |
| 3 | M | 88 | F508del/F508del | Hepatomegaly Jaundice Collateral circulation | ↑ Persistent aspartate aminotransferase/alanine aminotransferase | Hepatomegaly ↑ of hepatic echogenicity Lobular liver | Liver steatosis | Normal |
| 4 | F | 4 | Not found | Absent | Normal | Biliary lithiasis | – | – |
| 5 | F | 3 | Not found | Hepatomegaly Jaundice/Choluria Fecal acolia | ↑ Transient aspartate aminotransferase/alanine aminotransferase/gamma-glutamyl transferase | Normal | Intrahepatic cholestasis Bile in portal space | – |
| 6 | F | 3 | F508del/F508del | Hepatomegaly | ↑ Persistent Alkaline Phosphatase | Hepatomegaly | – | – |
| 7 | F | 59 | F 508del/3120+1G-A | Hepatomegaly | ↑ Persistent Alkaline Phosphatase ↓ Persistent prothrombin time | Hepatomegaly ↑ of hepatic echogenicity Liver steatosis | Fibrosis portal/septal and presinusoidal | Normal |
| 8 | F | 3 | G542X/R1162X | Hepatomegaly Jaundice/Choluria Fecal acolia Collateral circulation | ↑ Persistent aspartate aminotransferase/alanine aminotransferase Alkaline Phosphatase/gamma-glutamyl transferase/direct bilirubin ↓ Persistent prothrombin time/Albumin | Hepatomegaly ↑ of hepatic echogenicity Nodular liver and irregular border Portal hypertension | Biliary cirrhosis | – |
| 9 | F | 7 | F508del/F508del | Hepatomegaly | ↑ Persistent aspartate aminotransferase/alanine aminotransferase/Alkaline Phosphatase | Hepatomegaly ↑ of hepatic echogenicity Hepatic steatosis | – | Normal |
The present study demonstrated that 16.4% of the patients with CF had hepatobiliary disease. This frequency is similar to that described by Wagener et al. (16.3%), who evaluated 30,727 CF patients from a multicenter study in the United States and Canada between 1999 and 2004.10 However, Fagundes et al. followed-up 106 Brazilian patients with CF for approximately one year and found a lower frequency (9.4%) of hepatobiliary disease associated with CF.2 In addition to the short follow-up period, those authors used only biochemical and/or clinical alterations as diagnostic criteria, which may have led to the underdiagnosis of this complication.
The prevalence of CF-associated hepatobiliary disease is quite variable in the literature, due to the heterogeneous clinical presentation, absence of universal diagnostic criteria, and the variation of characteristics inherent to the population (age, gender, survival rate).17 Longitudinal studies in Europeans, North-Americans, and Brazilians have shown prevalence rates varying between 5.7% and 39%.4–11
Male gender has been considered a risk factor for the development of hepatobiliary disease associated with CF. Colombo et al. demonstrated a 2.5-fold higher risk of hepatobiliary disease in males.7 However, the present study showed a higher frequency of males in the group without hepatobiliary disease (p=0.042). The predominance of females among the patients with hepatobiliary disease (77.8%) is in agreement with data from the study by Lindblad et al., in which 68.6% of patients with CF and hepatopathy were females.6
In this study, the age at the diagnosis of hepatobiliary disease associated with CF was early (median of 7 months), ranging from 3 months to 3 years and 11 months; hepatobiliary manifestations were among the first signs and symptoms of CF in 55.6% (5/9) of these patients. Additionally, it is important to emphasize that the diagnosis of hepatobiliary disease occurred before and/or concomitantly with that of CF in 88.9% (8/9) of them.
However, Ciucã et al., when studying 66 Romanians with CF, demonstrated that only three patients (4.5%) had hepatobiliary disease as the first manifestation of CF, with a mean interval of four years between the CF diagnosis and hepatopathy.18 Other studies have also demonstrated a later onset of hepatobiliary disease, ranging from 3 to 8 years.2,7–9,18,19 Therefore, the findings of the present study demonstrate the early onset of this manifestation in this population of patients with CF and the importance of investigating CF in patients with hepatopathy.
Among the possible justifications for the early presentation is the fact that patients with hepatobiliary disease have more severe CF phenotypes, which was corroborated by the association between the presence of severe pancreatic insufficiency (88.9% vs. 31.6%; p=0.007) and severe mutations in the CFTR gene (77.8% vs. 39.6%, p=0.033) in this group, compared to those without hepatobiliary disease.
Moreover, Lamireau et al. demonstrated that pancreatic insufficiency is an independent factor associated with an increased prevalence of hepatobiliary disease in CF, leading to a 9.8-fold higher risk of liver disease in patients with CF.8 Although the literature does not describe an association between any mutation related to the CFTR gene and the presence or severity of hepatobiliary disease, some studies have demonstrated that more severe genotypes may be considered risk factors.20,21
Cirrhosis occurs predominantly in individuals with Class I, II, and III mutations.1,10,22,23 A study of 174 Romanians with CF disclosed a five-fold higher risk of developing hepatobiliary disease in patients with severe mutations (OR: 4.79), which were identified in 78% of those with hepatobiliary disease.18 However, it is known that liver disease development is very variable among patients with the same CFTR mutations, suggesting a possible multigenic role of the genetic polymorphism and environmental factors, not evaluated in the study, in the pathogenesis of this complication.1,3,13,21,24 Therefore, later studies may assess the role of these variables in the development of hepatobiliary disease.
Poor lung function and nutritional status indicators in CF have also been studied as risk factors for hepatobiliary disease, and could justify the severity and early onset of this complication. Fagundes et al. identified an association between W/A Z-score <−2 and development of hepatobiliary disease; these authors also observed that Shwachman–Kulczycki score values ≤70 conferred a 7.5-fold higher risk of developing this complication in patients with CF.25 In the present study, 44.4% of the patients with hepatobiliary disease presented Shwachman–Kulczycki score ≤70, and some degree of nutritional impairment was observed in 66.7% of them. However, it is necessary to compare these variables with the group without hepatobiliary disease to reinforce the importance of these findings.
Although not statistically significant, it is worth mentioning that the CF diagnosis was attained much earlier in the group with hepatobiliary disease, when compared to those without this complication (median of 7 vs. 59 months, p=0.059). Similarly, Fagundes et al. demonstrated that the age at CF diagnosis was independently associated with the development of liver disease, being inversely proportional.25
It is possible that hepatobiliary disease is part of a spectrum of worse prognosis and severity of CF, which would justify the earlier disease diagnosis in these patients. It is possible that a larger sample could demonstrate this association, since this study evaluated only 55 children.
Liver disease with cirrhosis and its complications tend to occur in approximately 7–12% of CF patients between the ages of 7 and 10.6,18,19 Lamireau et al. demonstrated a median time of 10 years for the evolution of hepatopathy associated with CF to portal hypertension. In the present study, only two patients with CF (3.6%), who already had portal hypertension since the diagnosis, presented liver cirrhosis at 5 months and 3 years of age. Non-performance of hepatic biopsy in all patients and the young age of children with hepatobiliary disease (median of 7 months) may justify the lower frequency of cirrhosis, since it is a late consequence, secondary to progressive hepatocyte lesion due to biliary thickening and its toxicity.17
However, it is noteworthy the precocity of cirrhosis and portal hypertension onset in the present study, suggesting a more severe spectrum of liver disease in this population. A possible justification would be the presence of concomitant CMV infection, worsening the liver status. This infection occurs in 50% of the children in the first year of life in places with high seropositivity rates,26 which is associated with precarious socioeconomic conditions (found in the studied population). However, this association has not been confirmed. In turn, although this infection was investigated in all patients through serology, only two were identified using the PCR technique and, thus, underdiagnosis may have occurred.
Among the study limitations, the authors emphasize the small sample of patients with CF and the number of patients with hepatobiliary disease. Moreover, only four patients underwent a liver biopsy, which may have underestimated the occurrence of liver disease. Additionally, it was not possible to confirm the genetic diagnosis of CF in all patients, since only seven mutations were investigated. Furthermore, the role of genetic polymorphisms in the development of this complication was not evaluated. Nonetheless, the results were significant.
In conclusion, this study found a high frequency of hepatobiliary disease in patients with CF (16.4%), which occurred as an initial manifestation of the latter in 55.6% of patients with liver disease. It is worth mentioning the earlier occurrence of hepatobiliary disease, as well as the early onset of complications in relation to the literature. This emphasizes the importance of including CF in the differential diagnosis of all liver diseases in childhood. Additionally, an association was observed between the presence of pancreatic insufficiency and severe mutations in the CFTR gene and the occurrence of hepatobiliary disease. However, further studies involving a larger number of patients should be carried out, aiming to better assess other risk factors for the development of hepatobiliary disease in CF.
FundingPartial funding from Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) under No. 020/2013 – Programa de Pesquisa para o SUS da Bahia: gestão partilhada de saúde – FAPESB.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Nascimento FS, Sena NA, Ferreira TA, Marques CD, Silva LR, Souza EL. Hepatobiliary disease in children and adolescents with cystic fibrosis. J Pediatr (Rio J). 2018;94:504–10.
Study carried out at Universidade Federal da Bahia (UFBA), Salvador, BA, Brazil.




