


The aims of the study were to determine the frequency of hepatobiliary disease in patients with cystic fibrosis and to describe the sociodemographic, clinical, and laboratory profile of these patients.
MethodsThis was a retrospective, descriptive, and analytical study of 55 patients diagnosed with cystic fibrosis, aged between 3 months and 21 years, followed‐up from January 2008 to June 2016 in a referral center. Medical records were consulted and sociodemographic, clinical and laboratory data, including hepatobiliary alterations, imaging studies, genetic studies, liver biopsies, and upper digestive endoscopies were registered.
ResultsHepatobiliary disease was diagnosed in 16.4% of the patients and occurred as an initial manifestation of cystic fibrosis in 55.6% of these cases. The diagnosis of hepatopathy occurred before or concomitantly with the diagnosis of cystic fibrosis in 88.9% of the children. All patients with hepatobiliary disease were considered non‐white, with a predominance of females (77.8%) and median (IQR) of 54 (27–91) months. Compared with the group without hepatobiliary disease, children with liver disease had a higher frequency of severe mutations identified in the CFTR gene (77.8% vs. 39.6%, p=0.033) and severe pancreatic insufficiency (88.9% vs. 31.6%, p=0.007).
ConclusionThe frequency of hepatobiliary disease was high, with a very early diagnosis of the disease and its complications in the studied series. A statistical association was observed between the occurrence of hepatobiliary disease and the presence of pancreatic insufficiency and severe mutations in the CFTR gene. It is emphasized that cystic fibrosis is an important differential diagnosis of liver diseases in childhood.
Os objetivos do estudo foram determinar a frequência da doença hepatobiliar em pacientes com fibrose cística e descrever o perfil sociodemográfico, clínico e laboratorial destes.
MétodosEstudo retrospectivo, descritivo e analítico de 55 pacientes com diagnóstico de fibrose cística, entre três meses e 21 anos, acompanhados de janeiro de 2008 a junho de 2016 em um centro de referência. Foi realizada consulta aos prontuários médicos, registrando‐se os dados sociodemográficos, clínicos e laboratoriais, incluindo‐se alterações hepatobiliares, exames de imagem, estudos genéticos, biópsias hepáticas e endoscopias digestivas altas.
ResultadosA doença hepatobiliar foi diagnosticada em 16,4% dos pacientes e ocorreu como manifestação inicial da fibrose cística em 55,6% destes casos. O diagnóstico da hepatopatia ocorreu antes ou concomitantemente ao diagnóstico da fibrose cística em 88,9% das crianças. Todos os pacientes com doença hepatobiliar foram considerados não brancos, havendo predominância do sexo feminino (77,8%) e mediana (I.I.Q) de idade de 54 (27‐91) meses. Em comparação com o grupo sem doença hepatobiliar, as crianças com hepatopatia tiveram maior frequência de mutações graves no gene CFTR identificadas (77,8% vs. 39,6%; p=0,033) e de insuficiência pancreática grave (88,9% vs. 31,6%; p=0,007).
ConclusãoA frequência de doença hepatobiliar foi elevada, observou‐se um diagnóstico muito precoce da mesma e de suas complicações na casuística estudada. Houve associação estatística entre a ocorrência de doença hepatobiliar e a presença de insuficiência pancreática e de mutações graves do gene CFTR. Enfatiza‐se que a fibrose cística represente um importante diagnóstico diferencial de hepatopatias na infância.
A fibrose cística (FC) é a doença genética letal mais comum nos brancos. O transporte anormal de cloro e sódio através da proteína reguladora de condutância transmembrana (CFTR) defeituosa causa um aumento na densidade das secreções exócrinas, promove o acometimento de uma série de órgãos.1
As principais manifestações clínicas da FC concentram‐se nos tratos respiratório e gastrointestinal. Entretanto, com o aumento da expectativa de vida dos pacientes com FC nas últimas décadas, a doença hepatobiliar associada à FC tem aumentado sua prevalência e ganhado mais notoriedade entre pacientes, parentes e profissionais de saúde.2,3
Habitualmente, as manifestações hepatobiliares associadas à FC se iniciam no fim da primeira década de vida dos pacientes, com prevalência de 5,7% a 39%.4–11 A doença hepatobiliar é a terceira causa de mortalidade nos pacientes com FC, precedida pelas mortes relacionadas ao acometimento pulmonar e às complicações inerentes aos transplantes de órgãos.12 Além disso, alguns estudos têm demonstrado que pacientes com doença hepática avançada estão em risco de desenvolver e/ou agravar outras manifestações extra‐hepáticas da FC, como desnutrição, diabetes mellitus, osteodistrofia hepática e quadro pulmonar, acarreta maior morbidade pela doença de base.3,13 Dessa forma, a doença hepatobiliar tem sido considerada um fator preditor de risco na evolução e no prognóstico desfavoráveis da FC.14
Poucos estudos foram feitos sobre a doença hepatobiliar relacionada à FC na população brasileira, é fundamental conhecer as particularidades no Brasil, que tem características étnicas peculiares nas diferentes regiões geográficas. Os objetivos do estudo foram determinar a frequência da doença hepatobiliar em pacientes com FC e descrever o perfil sociodemográfico, clínico e laboratorial desses.
MétodosEstudo retrospectivo, descritivo e analítico de pacientes atendidos em um centro de referência de FC em um hospital universitário. Foram analisados os prontuários de todos os pacientes, de três meses a 21 anos, acompanhados entre janeiro de 2008 e junho de 2016, com diagnóstico de FC confirmado por dois testes do suor positivos e/ou estudo genético, que tivessem pelo menos três meses de seguimento no serviço. Foram excluídos os pacientes que apresentassem comorbidades que afetassem o sistema hepatobiliar (uso de drogas hepatotóxicas e/ou doenças hepatobiliares primárias ou que cursassem secundariamente com acometimento do fígado, vesícula e/ou baço).
A consulta aos prontuários foi feita entre junho e julho de 2016 e obtiveram‐se características sociodemográficas, clínicas – idade (meses), peso (kg), altura (cm) e índice de massa corpórea (IMC) ao diagnóstico da FC; tempo de seguimento no centro; presença de manifestações: hepatobiliares (icterícia, hepatomegalia, esplenomegalia, circulação colateral), pulmonares (pneumonias de repetição, sibilância, colonização por Pseudomonas aeruginosa) e gastrointestinais (diarreia, esteatorreia, baixo ganho ponderal, íleo meconial); escore de Swachaman‐Kulczycki15 – exames laboratoriais/imagem e estudo genético. Informações de exames histopatológicos (biópsia hepática) e endoscopia digestiva alta (EDA) foram registradas, quando presentes. O peso, a altura e o IMC foram avaliados pelo escore‐z, conforme a classificação da Organização Mundial de Saúde (OMS, 2006). Pacientes com elastase fecal < 100μg/g foram classificados com insuficiência pancreática grave. A ultrassonografia (USG) de abdome foi feita anualmente nos pacientes maiores de sete anos ou mais precocemente naqueles que apresentassem sinais ou sintomas ou alterações das provas laboratoriais sugestivos de doença hepatobiliar.
A idade do diagnóstico da FC foi definida pela época do resultado do segundo teste do suor positivo ou do estudo genético. O tempo de seguimento de cada paciente correspondeu ao período entre junho de 2016 e a primeira consulta ou primeiro internamento no serviço. A cor da pele foi definida por um dos pesquisadores, coordenador médico do ambulatório multidisciplinar de fibrose cística, com base nas características fenotípicas dos pacientes e de seus pais.
Todos os pacientes foram submetidos ao estudo genético, pesquisaram‐se, através das técnicas de reação em cadeia da polimerase (PCR) ou digestão enzimática,16 as seguintes mutações no gene CFTR: F508del, G542X, R1162X, R334W, 3120+1G‐A, G551D e R553X. A infecção pelo vírus citomegalovírus (CMV) foi pesquisada através de sorologias (anticorpos IgM e IgG), nos pacientes com critérios para doença hepatobiliar, foi também feita a PCR, quando disponível.
A doença hepatobiliar, associada à FC, foi definida por: alterações compatíveis à biópsia hepática ou evidência de colangiopatias (microvesícula, litíase biliar ou estenose de ductos biliares) ou presença, por três meses consecutivos, de pelo menos dois dos critérios: (1) hepatomegalia ao exame físico (fígado > 2cm do rebordo costal direito, confirmada por USG de abdome); (2) elevação acima do limite superior de normalidade para aspartato aminotransferase e/ou alanina aminotransferase e/ou gama glutamiltransferase; (3) alteração na USG de abdome (aumento da ecogenicidade hepática, nodularidade, borda hepática irregular, fibrose hepática ou sinais de hipertensão portal).7 A idade de diagnóstico da doença hepatobiliar foi determinada no momento em que algum critério fosse observado.
Para a elaboração da base de dados e análise estatística, foi usado o software SPSS® (IBM SPSS Statistics para Windows, versão 20.0, NY, EUA). Foi feita análise descritiva das características sociodemográficas, clínicas e laboratoriais, caracterizadas por medidas de tendência central e dispersão (médias e desvio‐padrão, mediana e intervalo interquartil). Foi feita comparação dos grupos com e sem doença hepatobiliar através do teste de Mann‐Whitney para variáveis numéricas e do teste exato de Fisher para as variáveis categóricas. Valores de p ≤ 0,05 foram considerados estatisticamente significantes em todas as análises.
O estudo foi aprovado pelo Comitê de Ética e Pesquisa da instituição, registro 121/2011. Todos os participantes e seus responsáveis receberam informações verbais e por escrito sobre a natureza, a proposta e o objetivo do estudo e possíveis riscos e benefícios e deram por escrito o seu consentimento informado.
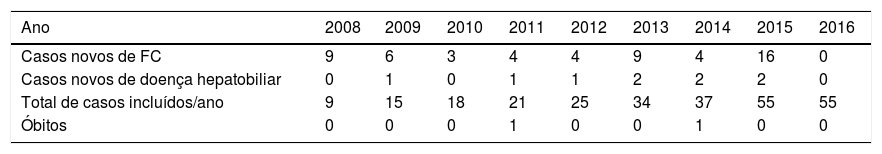
ResultadosForam analisados 55 pacientes com FC. A mediana (I.I.Q) do tempo de seguimento desses pacientes foi de 35 (15 a 78) meses, variou de três a 102 meses. A distribuição do número de casos novos de FC e de doença hepatobiliar, por ano de seguimento, está descrita na tabela 1.
Número de casos novos de fibrose cística e de doença hepatobiliar, óbitos e total de casos acompanhados entre janeiro de 2008 a junho de 2016 em um centro de referência
| Ano | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 |
|---|---|---|---|---|---|---|---|---|---|
| Casos novos de FC | 9 | 6 | 3 | 4 | 4 | 9 | 4 | 16 | 0 |
| Casos novos de doença hepatobiliar | 0 | 1 | 0 | 1 | 1 | 2 | 2 | 2 | 0 |
| Total de casos incluídos/ano | 9 | 15 | 18 | 21 | 25 | 34 | 37 | 55 | 55 |
| Óbitos | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
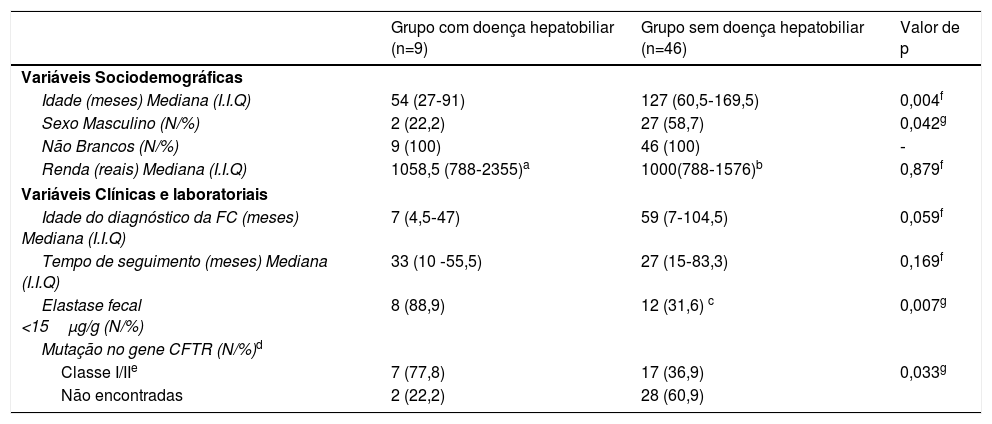
A doença hepatobiliar associada à FC foi diagnosticada em 16,4% (9/55) dos pacientes com FC. A mediana de idade (I.I.Q) desses pacientes foi de 54 (27 a 91) meses, variou de dez meses a nove anos e 11 meses. Sete (77,8%) pacientes eram do sexo feminino e todos foram considerados não brancos. As demais características sociodemográficas, clínicas e laboratoriais do grupo de pacientes com FC com e sem doença hepatobiliar estão descritas na tabela 2.
Características sociodemográficas, clínicas e laboratoriais dos pacientes com fibrose cística, acompanhados entre janeiro de 2008 a junho de 2016, em um centro de referência
| Grupo com doença hepatobiliar (n=9) | Grupo sem doença hepatobiliar (n=46) | Valor de p | |
|---|---|---|---|
| Variáveis Sociodemográficas | |||
| Idade (meses) Mediana (I.I.Q) | 54 (27‐91) | 127 (60,5‐169,5) | 0,004f |
| Sexo Masculino (N/%) | 2 (22,2) | 27 (58,7) | 0,042g |
| Não Brancos (N/%) | 9 (100) | 46 (100) | ‐ |
| Renda (reais) Mediana (I.I.Q) | 1058,5 (788‐2355)a | 1000(788‐1576)b | 0,879f |
| Variáveis Clínicas e laboratoriais | |||
| Idade do diagnóstico da FC (meses) Mediana (I.I.Q) | 7 (4,5‐47) | 59 (7‐104,5) | 0,059f |
| Tempo de seguimento (meses) Mediana (I.I.Q) | 33 (10 ‐55,5) | 27 (15‐83,3) | 0,169f |
| Elastase fecal <15μg/g (N/%) | 8 (88,9) | 12 (31,6) c | 0,007g |
| Mutação no gene CFTR (N/%)d | |||
| Classe I/IIe | 7 (77,8) | 17 (36,9) | 0,033g |
| Não encontradas | 2 (22,2) | 28 (60,9) | |
I.I.Q, Intervalo Interquartil; N, número de pacientes.
A idade, em mediana (I.I.Q), do diagnóstico da doença hepatobiliar associada à FC foi sete meses (três a 47). Esse diagnóstico ocorreu antes e/ou concomitante ao da FC em 88,9% dos pacientes, a presença de manifestações hepatobiliares estava entre os primeiros sinais e sintomas da FC em cinco dos nove pacientes (55,6%).
Os pacientes com hepatopatia tiveram maior frequência de mutações graves no gene CFTR (77,8% vs. 39,6%; p = 0,033) e de insuficiência pancreática grave (88,9% vs. 31,6%; p = 0,007) do que o grupo sem doença hepatobiliar. Embora não tenha sido estatisticamente significante, as crianças com doença hepatobiliar apresentaram diagnóstico da FC muito mais precoce em comparação com o segundo grupo, com mediana (I.I.Q) de sete (4,5 a 47) vs. 59 (sete a 104,5) meses, respectivamente (p = 0,059) (tabela 2).
Em relação às características clínicas, dos nove pacientes com doença hepatobiliar associada à FC, nenhum teve íleo meconial e um apresentava função pancreática normal com doença biliar assintomática. Os demais (88,9%) tinham insuficiência pancreática grave.
A tabela 3 apresenta as características clínicas no momento do diagnóstico da FC dos pacientes com doença hepatobiliar. Algum agravo nutricional (escore‐z peso/idade e/ou altura/idade e/ou IMC/ idade < ‐2) foi encontrado em seis (66,7%) desses pacientes. As medianas do escore‐z para peso/idade e altura/idade nesse grupo de pacientes foi de ‐3,27 e ‐3,08, com valores mínimos de ‐9,49 e ‐6,82, respectivamente. Os valores de escore‐z de IMC/idade foram > ‐2 em 77,8% dos casos, devido à redução proporcional dos valores de peso e altura (tabela 3).
Características clínicas no momento do diagnóstico da FC dos pacientes com doença hepatobiliar, acompanhados entre janeiro de 2008 a junho de 2016, em um centro de referência
| Z score Peso/Idade (mediana/I.I.Q) | −3,27 (−4,34 a −0,26) |
| Z score Altura/Idade (mediana/I.I.Q) | −3,08 (−4,4 a −0,46) |
| Z score Peso/Altura (mediana/I.I.Q) | −0,43 (−2,39 a 0,41) |
| Z score IMC/ Idade (mediana/I.I.Q) | −0,26 (1,86 a −0,15) |
| Manifestações pulmonares (N/%) | 9 (100) |
| Valor do Escore de Shwachman‐Kulczycki (mediana/I.I.Q) | 85 (57,5 a 87,5) |
| Classificação do Escore de Shwachman‐Kulczycki (N/%)a | |
| Excelente ou bom (100‐71) | 5 (66,6) |
| Médio (70‐56) | 1 (11,1) |
| Moderado ou Grave (≤ 55) | 3 (33,3) |
I.I.Q, Intervalo Interquartil; N, número de pacientes.
A forma de apresentação da doença hepatobiliar associada à FC está descrita na tabela 4. A hepatomegalia foi o achado mais encontrado, estava presente em sete pacientes (77,8%), seguida de aumento da ecogenicidade hepática em cinco crianças (55,5%). Todos os pacientes fizeram sorologia para citomegalovírus (CMV), a qual foi positiva em dois casos, um desses também confirmado por técnica de PCR. Um paciente com sorologia negativa teve a infecção por CMV confirmada por PCR. Esses pacientes apresentavam achados de biópsia hepática compatíveis com doença hepatobiliar da FC. Em relação às complicações da hepatopatia, foi observada hipertensão portal em dois pacientes, esplenomegalia em apenas um caso e varizes esofágicas em um dos quatro pacientes que fizeram EDA. Somente uma criança evoluiu com sinais de insuficiência hepática aguda.
Apresentação clínica dos pacientes com doença hepatobiliar associada à fibrose cística de um centro de referência, entre janeiro de 2008 e junho de 2016
| Caso | Sexo | Início da doença hepatobiliara | Genótipo | Clínica | Laboratório | USG de abdome | Biópsia hepática | EDA |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 19 | F508del/ 3120+1G‐A | Hepatomegalia | ↑Persistente AST/ GGT | Hepatomegalia | – | – |
| 2 | F | 35 | F508del/ G542X | Hepatomegalia Circulação colateral | ↑ Persistente FA ↓ Persistente TP | Hepatoesplenomegalia ↑ da ecogenicidade hepática Microvesicula Hipertensão portal | Cirrose bili | Varizes esofágicas |
| 3 | M | 88 | F508del/ F508del | Hepatomegalia Icterícia Circulação colateral | ↑Persistente AST/ALT | Hepatomegalia ↑ da ecogenicidade hepática Fígado lobular | Esteatose hepática | Normal |
| 4 | F | 4 | Não encontrada | Ausente | Normal | Litíase biliar | – | – |
| 5 | F | 3 | Não encontrada | Hepatomegalia Icterícia/ Colúria Acolia fecal | ↑ Transitória AST/ALT/GGT | Normal | Colestase intrahepática Bile no espaço porta | – |
| 6 | F | 3 | F508del/ F508del | Hepatomegalia | ↑ Persistente FA | Hepatomegalia | – | – |
| 7 | F | 59 | F 508del/ 3120+1G‐A | Hepatomegalia | ↑ Persistente FA ↓ Persistente TP | Hepatomegalia ↑ da ecogenicidade hepática Esteatose hepática | Fibrose portal/septal e perissinusiodal | Normal |
| 8 | F | 3 | G542X/ R1162X | Hepatomegalia Icterícia/ Colúria Acolia fecal Circulação colateral | ↑ Persistente AST/ALT FA/GGT/ BD ↓ Persistente TP/Albumina | Hepatomegalia ↑ da ecogenicidade hepática Fígado nodular e borda irregular Hipertensão portal | Cirrose biliar | – |
| 9 | F | 7 | F508del/ F508del | Hepatomegalia | ↑ Persistente AST/ALT/FA | Hepatomegalia ↑ da ecogenicidade hepática Esteatose hepática | – | Normal |
ALT, Alanina Aminotransferase; AST, Aspartato Aminotransferase; BD, Bilirrubina Direta; EDA, Endoscopia Digestiva Alta; F, Feminino; FA, Fosfatase Alcalina; GGT, Gamaglutamil Transferase; M, Masculino; TP, Tempo de Protrombina; USG, Ultrassonografia.
O presente estudo demonstrou que 16,4% dos pacientes com FC apresentaram doença hepatobiliar. Essa frequência foi semelhante à descrita por Wagener et al. (16,3%), que avaliaram 30.727 pacientes com FC de um estudo multicêntrico nos Estados Unidos e Canadá, entre 1999‐2004.11 No entanto, Fagundes et al. acompanharam 106 pacientes brasileiros com FC por cerca de um ano e encontraram frequência menor (9,4%) de doença hepatobiliar associada à FC.2 Além do curto período de seguimento, os autores usaram apenas alterações bioquímicas e/ou clínicas como critérios diagnósticos, o que pode ter levado ao não diagnóstico dessa complicação. A prevalência da doença hepatobiliar associada à FC é bastante variável na literatura, devido à apresentação clínica heterogênea, à ausência de critérios diagnósticos universais e à variação das características inerentes à população (idade, sexo, taxa de sobrevida).17 Alguns estudos longitudinais
em europeus, americanos e brasileiros têm mostrado prevalência entre 5,7% e 39%.4–11
O sexo masculino tem sido considerado um fator de risco para o desenvolvimento de doença hepatobiliar associada à FC. Colombo et al. demonstraram risco 2,5 vezes maior de doença hepatobiliar no sexo masculino.7 Porém, o presente estudo evidenciou maior frequência do sexo masculino no grupo sem doença hepatobiliar (p = 0,042). A predominância do sexo feminino nos pacientes com doença hepatobiliar (77,8%) concordou com os dados de Lindblad et al., nos quais 68,6% dos pacientes com FC e hepatopatia eram do sexo feminino.6
Neste estudo, a idade do diagnóstico da doença hepatobiliar associada à FC foi precoce (mediana de sete meses), variou de três meses a três anos e 11 meses, as manifestações hepatobiliares estavam entre os primeiros sinais e sintomas da FC em 55,6% (5/9) desses pacientes. Adicionalmente, é importante destacar que o diagnóstico da doença hepatobiliar ocorreu antes e/ou concomitante ao da FC em 88,9% (8/9) deles. No entanto, Ciucã et al., ao estudar 66 romenos com FC, demonstraram que apenas três pacientes (4,5%) apresentaram doença hepatobiliar como primeira manifestação da FC, com um intervalo médio de quatro anos entre o diagnóstico da FC e da hepatopatia.18 Outros trabalhos também têm demonstrado um surgimento mais tardio da doença hepatobiliar, variou entre três e oito anos2,7–9,18,19. Logo, os achados do presente estudo demonstram a precocidade dessa manifestação nessa população de pacientes com FC e a importância de se investigar FC em pacientes com hepatopatia.
Entre as possíveis justificativas para a apresentação tão precoce está o fato dos pacientes com doença hepatobiliar terem fenótipos mais graves da FC, o que foi corroborado pela associação demonstrada entre a presença de insuficiência pancreática grave (88,9% vs. 31,6%; p = 0,007) e mutações graves no gene CFTR(77,8% vs. 39,6%; p = 0,033) nesse grupo, comparado ao sem doença hepatobiliar.
Adicionalmente, Lamireau et al. demonstraram que a insuficiência pancreática é um fator independente associado ao aumento da prevalência da doença hepatobiliar na FC, leva a um risco 9,8 vezes maior de hepatopatia nos pacientes com FC.8 Apesar de a literatura não descrever associação entre qualquer mutação relacionada ao CFTR e a presença ou gravidade da doença hepatobiliar, alguns estudos têm mostrado que genótipos mais graves podem ser considerados fatores de risco.20,21 A cirrose ocorre predominantemente em indivíduos com mutações das classes I, II e III.1,10,22,23 Estudo feito com 174 romenos com FC evidenciou risco cerca de cinco vezes maior de desenvolver doença hepatobiliar em pacientes com mutações graves (OR: 4,79), as quais foram identificadas em 78% daqueles com doença hepatobiliar.18 Entretanto, sabe‐se que o desenvolvimento doença hepática é muito variável entre pacientes com mesmas mutações no CFTR, sugere um possível papel multigênico, do polimorfismo genético e de fatores ambientais, não avaliado no estudo, na patogênese dessa complicação.1,3,13,21,24 Assim, estudos posteriores poderão avaliar o papel dessas variáveis no desenvolvimento da doença hepatobiliar.
Indicadores de função pulmonar e do estado nutricional ruins na FC também têm sido estudados como fatores de risco para doença hepatobiliar e poderiam justificar a gravidade e precocidade dessa complicação. Fagundes et al. identificaram uma associação entre escore‐z de P/I < ‐2 e desenvolvimento de doença hepatobiliar e que valores de escore de Shwachman‐Kulczycki ≤ 70 conferiam um risco 7,5 vezes maior de desenvolvimento dessa complicação nos pacientes com FC.25 No presente estudo, 44,4% dos pacientes com doença hepatobiliar tinha esse escore ≤ 70 e havia algum agravo nutricional em 66,7% deles. Porém, torna‐se necessária a comparação dessas variáveis com o grupo sem doença hepatobiliar para fortalecer a importância desses achados.
Embora não tenha sido estatisticamente significante, vale ressaltar que o diagnóstico da FC foi muito mais precoce no grupo com doença hepatobiliar comparado com aqueles sem essa complicação (mediana de sete vs. 59 meses; p = 0,059). Em concordância, Fagundes et al. demonstraram que a idade ao diagnóstico da FC estava independentemente associada ao desenvolvimento de hepatopatia, de maneira inversamente proporcional.25 É possível que a doença hepatobiliar faça parte de um espectro de pior prognóstico e gravidade da FC, o que justificaria o diagnóstico mais precoce da doença nesses pacientes. É possível que uma amostra de maior tamanho pudesse demonstrar essa associação, desde que este estudo avaliou apenas 55 crianças.
A doença hepática com cirrose e suas complicações tendem a ocorrer em cerca de 7% a 12% dos pacientes com FC, entre sete e 10 anos.6,18,19 Lamireau et al. demonstraram um tempo mediano de 10 anos para a evolução da hepatopatia associada a FC à hipertensão portal. No presente estudo, apenas dois pacientes com FC (3,6%), os quais já tinham hipertensão portal desde o diagnóstico, apresentaram cirrose hepática, aos cinco meses e três anos. A não feitura de biópsia hepática em todos os pacientes e a idade precoce das crianças com doença hepatobiliar (mediana de sete meses) podem justificar a menor frequência de cirrose, já que é uma consequência tardia, secundária à lesão progressiva de hepatócitos, devido ao espessamento biliar e à sua toxicidade.17 Porém, chama atenção a precocidade da cirrose e da hipertensão portal no atual trabalho, sugere um espectro mais grave da hepatopatia nessa população. Uma possível justificativa seria a presença de infecção concomitante pelo CMV, que agravou o quadro hepático. Essa infecção chega a ocorrer em 50% das crianças no primeiro ano de idade em locais com altas taxas de soropositividade,26 o que está associado às precárias condições socioeconômicas (presentes na população estudada). Entretanto, essa associação não foi confirmada. Por outro lado, embora essa infecção tenha sido investigada em todos os pacientes através da sorologia, apenas dois o fizeram pela técnica de PCR, pode ter ocorrido subdiagnóstico.
Dentre as limitações deste trabalho destaca‐se o pequeno tamanho da casuística de pacientes com FC e do número de pacientes com doença hepatobiliar. Também, apenas quatro pacientes fizeram biópsia hepática, o que pode ter subestimado a ocorrência da hepatopatia. Além disso, não foi possível confirmar o diagnóstico genético da FC em todos os pacientes, desde que apenas sete mutações foram pesquisadas. Ainda mais, não foi avaliado o papel do polimorfismo genético no desenvolvimento dessa complicação. Apesar disso, os resultados obtidos foram expressivos.
Este trabalho encontrou uma alta frequência de doença hepatobiliar entre os pacientes com FC (16,4%), que ocorreu como manifestação inicial da FC em 55,6% dos pacientes com hepatopatia. Destaca‐se a ocorrência mais precoce da doença hepatobiliar, bem como o aparecimento de complicações em relação à literatura. Isso enfatiza a importância de se incluir a FC no diagnóstico diferencial de todas as hepatopatias na infância. Além disso, foi observada associação entre presença de insuficiência pancreática e de mutações graves no gene CFTR e a ocorrência de doença hepatobiliar. No entanto, novos estudos que envolvam número maior de pacientes devem ser feitos, para avaliar melhor outros fatores de risco para o desenvolvimento da doença hepatobiliar na FC.
FinanciamentoFinanciamento parcial da Fundação de Amparo à Pesquisa do Estado da Bahia (Fabesp) sob o número 020/2013 – Programa Pesquisa para o SUS da Bahia: gestão partilhada de saúde.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Nascimento FS, Sena NA, Ferreira TA, Marques CD, Silva LR, Souza EL. Hepatobiliary disease in children and adolescents with cystic fibrosis. J Pediatr (Rio J). 2018;94:504–10.
Trabalho vinculado à Universidade Federal da Bahia (UFBA), Salvador, BA, Brasil.




