

To characterize the clinical, laboratory, and anthropometric profile of a sample of Brazilian patients with glycogen storage disease type I managed at an outpatient referral clinic for inborn errors of metabolism.
MethodsThis was a cross-sectional outpatient study based on a convenience sampling strategy. Data on diagnosis, management, anthropometric parameters, and follow-up were assessed.
ResultsTwenty-one patients were included (median age 10 years, range 1–25 years), all using uncooked cornstarch therapy. Median age at diagnosis was 7 months (range, 1–132 months), and 19 patients underwent liver biopsy for diagnostic confirmation. Overweight, short stature, hepatomegaly, and liver nodules were present in 16 of 21, four of 21, nine of 14, and three of 14 patients, respectively. A correlation was found between height-for-age and BMI-for-age Z-scores (r=0.561; p=0.008).
ConclusionsDiagnosis of glycogen storage disease type I is delayed in Brazil. Most patients undergo liver biopsy for diagnostic confirmation, even though the combination of a characteristic clinical presentation and molecular methods can provide a definitive diagnosis in a less invasive manner. Obesity is a side effect of cornstarch therapy, and appears to be associated with growth in these patients.
Caracterizar o perfil clínico, laboratorial e antropométrico de uma amostra de pacientes brasileiros com doença de depósito de glicogênio tipo I tratados em um ambulatório de referência para erros inatos do metabolismo.
MétodosEste foi um estudo ambulatorial transversal com base em uma estratégia de amostragem de conveniência. Foram avaliados os dados com relação ao diagnóstico, tratamento, parâmetros antropométricos e acompanhamento.
ResultadosForam incluídos 21 pacientes (idade média de 10 anos, faixa 1-25 anos de idade), e todos se encontravam em terapia de amido de milho cru. A idade média na época do diagnóstico foi de sete meses (faixa, 1-32 meses), e 19 pacientes foram submetidos a biópsia hepática para confirmação do diagnóstico. Sobrepeso, baixa estatura, hepatomegalia e nódulos hepáticos foram fatores presentes em 16 de 21, quatro de 21, nove de 14 e três de 14 pacientes, respectivamente. Foi encontrada uma correlação entre os escores z para peso para idade e IMC para idade (r=0,561; p=0,008).
ConclusõesO diagnóstico da doença de depósito de glicogênio tipo I tem sido tardio no Brasil. A maioria dos pacientes foi submetida a confirmação do diagnóstico, apesar de o quadro clínico característico e os métodos moleculares poderem fornecer um diagnóstico definitivo de forma menos invasiva. Obesidade é um efeito colateral da terapia com amido de milho e parece estar associada a crescimento nesses pacientes.
Glycogen storage disease type I (GSDI, or von Gierke's disease) is caused by deficiency of glucose-6-phosphatase (G6Pase), an enzyme that catalyzes hydrolysis of glucose-6-phosphate (G6P) into glucose and inorganic phosphate (Pi), a key step in the maintenance of glucose homeostasis. Two major subtypes of GSDI are recognized: GSD type Ia (GSDIa), which is the result of a mutation affecting the catalytic subunit of G6Pase-alpha (or G6PC), and GSD type Ib (GSDIb), which is caused by a defect in G6P translocase (or G6PT).1 GSDI is inherited in an autosomal recessive pattern, and its incidence is estimated at one in 100,000 live births, making it the most common of the hepatic GSDs.2
Patients with GSDIa present with hepatomegaly, a characteristic “doll-like” face, short stature, and chronic fatigue. Laboratory findings suggestive of GSDIa include hypoglycemia after a four to six hour fast, lactic acidosis, hypertriglyceridemia, and hyperuricemia. Functional tests for differential diagnosis of hypoglycemia show absence of glycemic response to glucagon injection and aggravation of hyperlactacidemia,3 whereas histopathological analysis of hepatic biopsy specimens shows glycogen buildup in the liver. In GSDIb, the clinical presentation is quite similar to that of GSDIa, but may be accompanied by neutropenia with recurrent infections (particularly of the gastrointestinal tract) and an increased incidence of inflammatory bowel disease.4 Although the gold-standard methods for diagnosis of GSDIa are the measurement of G6PC or G6PT activity in liver tissue and/or detection of pathogenic mutations in the genes that code for G6PC and G6PT, specific therapy can be initiated based solely on the clinical and histopathological findings.3 Access to the DNA/enzyme tests is limited since they are only provided by a select few national and international centers, usually within the framework of research projects.
Management of GSDI is essentially dietary,3 and consists of frequent meals – preferably containing slow-release carbohydrates such as uncooked cornstarch – at regular intervals, and restriction of fructose, sucrose, and lactose intake. In infants, the recommended management strategy includes frequent meals and continuous nocturnal infusion of glucose at a rate of 6-8mg/kg/min through a nasogastric or gastrostomy tube. Treatment efficacy is measured by monitoring growth and biochemical parameters, as well as by abdominal ultrasound for assessment of liver volume and presence of nodules. Proper dietary management decreases the risk of long-term complications, which include short stature, osteoporosis or bone mineral loss, kidney disease with hypertension, proteinuria, renal calculi, nephrocalcinosis, hepatocellular adenomas (with potential for malignant transformation), pancreatitis secondary to hypertriglyceridemia, and potentially life-threatening hypoglycemia.5,6
The objective of this study was to assess the clinical and laboratory profile of a sample of Brazilian patients with GSDI recruited from an outpatient referral center for inborn errors of metabolism. The main research hypothesis was that diagnosis of GSDI is delayed in Brazil, both due to a lack of access to diagnostic methods and due to poor awareness of the condition by healthcare providers, thus hindering early access to specific treatment and genetic counseling.
MethodsThis study was approved by the Ethics Committee of Hospital de Clínicas de Porto Alegre (HCPA, Brazil). All subjects signed an informed consent prior to study participation.
This was an outpatient-based case series with cross-sectional analysis of the variables of interest. A convenience sampling strategy was used. The study was conducted between March of 2011 and January of 2013. The criterion for inclusion was a diagnosis of GSDI established using at least two of the following methods (the diagnosis was independently confirmed by the authors in all patients): a) clinical diagnosis, defined by over 12 months of specialist care (led by hepatologist or medical geneticist) and clinical manifestations consistent with GSDI (hypoglycemia with hyperlactatemia, hypertriglyceridemia, hyperuricemia, hepatomegaly, and/or growth failure and short stature, and normal levels of creatine phosphokinase [CPK]) at the time of diagnosis or at the time of study inclusion; b) positive family history consistent with autosomal recessive inheritance, as long as GSDI had been confirmed by enzymatic methods or DNA analysis in the affected relative(s); c) histopathological diagnosis, defined as the presence of histological changes in liver tissue consistent with GSD, such as hyperglycogenated nuclei, mild fibrosis, and fatty changes with lipid vacuoles;7 d) enzymatic diagnosis, defined by negligible activity (< 10%) of G6Pase in fresh or frozen liver tissue samples; or e) molecular diagnosis, defined by the presence of pathogenic mutations in the G6PC gene (for patients with GSDIa) or in the SLC37A3 gene (for those with GSDIb) as detected by molecular methods. The distinction between GSDIa and GSDIb was mostly based on clinical findings (absence or presence of neutropenia, respectively), as molecular diagnostics were unavailable to the majority of patients.
Patients were invited to take part in the study after routine visits. Those who agreed to participate were all assessed by the same researcher and underwent a targeted history, physical examination, and anthropometric assessment. The latest laboratory values (blood glucose, lactate, cholesterol, triglycerides, uric acid) and imaging findings available for each patient were obtained by means of a chart review. Tests performed up to three months prior to anthropometric assessment were considered acceptable. The variables of interest were sex, parental consanguinity, current age, age at diagnosis (defined as the age at which parents reported a specific diagnosis of GSD or, if unavailable, the age at diagnosis as noted in the patient's first chart containing diagnostic test results and start of dietary management), first clinical manifestation (as reported by parents), laboratory parameters (current and at time of diagnosis), liver biopsy for histopathological examination or molecular analysis, and current clinical and imaging data (anthropometric assessment, liver ultrasound, and bone mineral density and body composition by dual-energy X-ray absorptiometry [DEXA]).
Anthropometric assessment consisted of weight (kg) and height (cm) measurement. Body weight was measured using digital scales with a maximum capacity of 150kg and a resolution of 100g, certified by the Brazilian National Institute of Metrology, Standardization, and Industrial Quality (Instituto Nacional de Metrologia, Qualidade e Tecnologia - INMETRO). Patients were weighed while nude and barefoot. Height was measured with a wall-mounted stadiometer precise to 1mm. In adolescents, the Tanner scale was used for pubertal staging. Anthropometric measurements and classifications for age and sex were calculated in the World Health Organization's AnthroPlus software suite. The variables of interest were height-for-age and BMI-for-age Z-scores, as proposed by the Brazilian Society of Pediatrics.8
Liver size was measured by ultrasonography and assessed for normality on the basis of the reference sizes for children published in 2010 by Dhingra et al.9 When objective data on liver size were missing, the sonographer's impression was used instead (normal or enlarged).
The criteria for adequacy of metabolic control were based on the European Study on Glycogen Storage Disease Type 1 (ESGSD I):5 blood glucose > 63mg/dL, triglycerides<530mg/dL, uric acid<7mg/dL, BMI between 0 and+2 standard deviations (SD), and lactate > 2.5 mmol/L (the latter used as the urine lactate/creatinine ratio was unavailable). The absence of hepatic adenomas and adequate height-for-age (z-score > -2 SD) are important parameters for assessment of metabolic control adequacy, but are not part of the ESGSD I.5
Statistical analyses were conducted in the Statistical Package for the Social Sciences® version 20.0 (SPSS Inc., Chicago, IL, USA). Continuous variables were expressed as means and standard deviations or as medians and interquartile ranges. Analysis of variance (ANOVA) was used for comparison of height and BMI z-scores. The significance level was set at 5%. Data were entered into a Microsoft Excel 2010 for Windows spreadsheet (Microsoft, Redmond, WA, EUA) and analyzed in SPSS 20.0 (IBM Corp., Armonk, NY, USA).
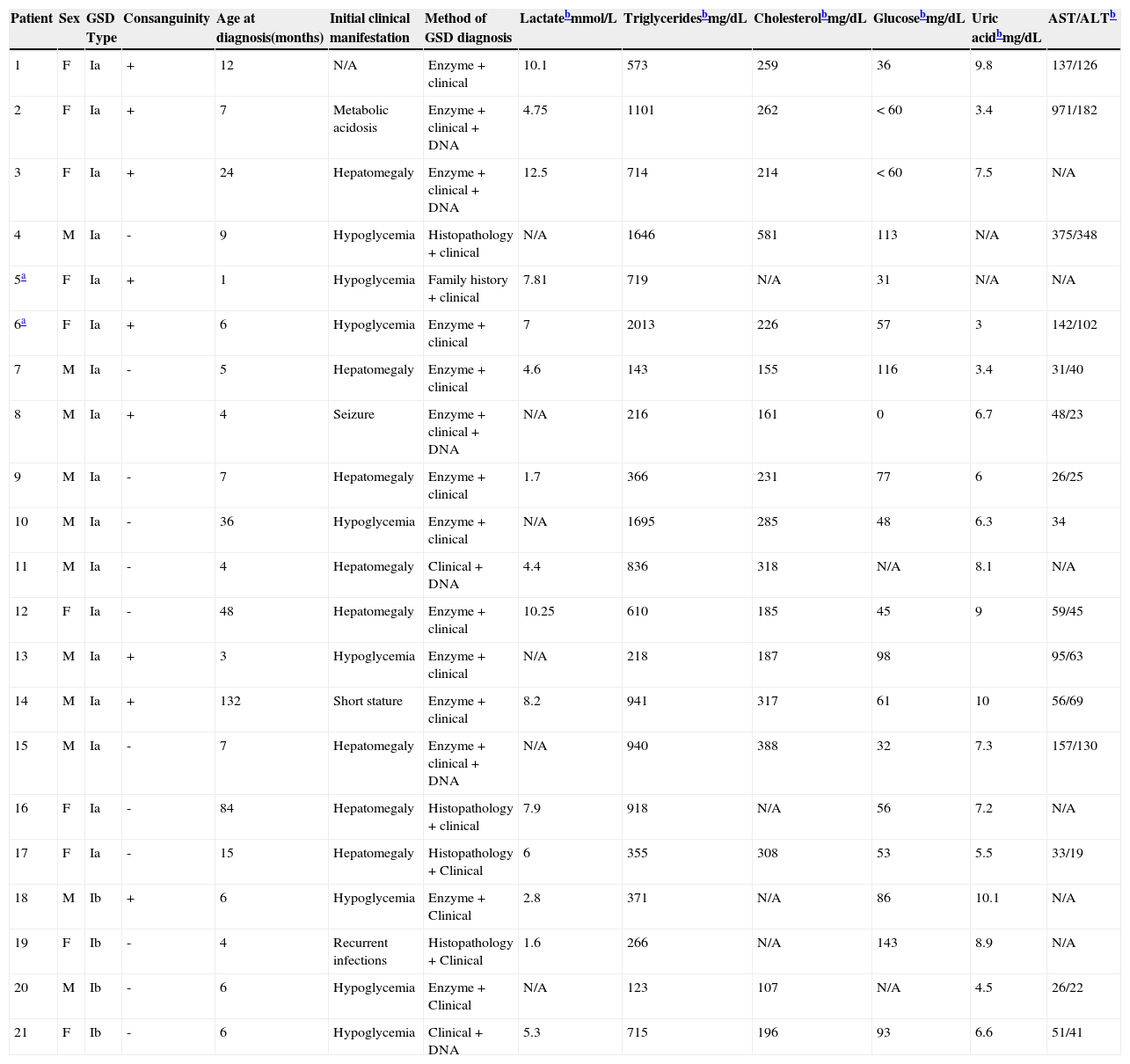
ResultsTwenty-one patients were included in the study: 17 with GSDIa and four with GSDIb. Table 1 shows the sample profile at the time of diagnosis.
Summary of findings at diagnosis among patients with glycogen storage disorder (GSD) type I (n=21).
| Patient | Sex | GSD Type | Consanguinity | Age at diagnosis(months) | Initial clinical manifestation | Method of GSD diagnosis | Lactatebmmol/L | Triglyceridesbmg/dL | Cholesterolbmg/dL | Glucosebmg/dL | Uric acidbmg/dL | AST/ALTb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Ia | + | 12 | N/A | Enzyme + clinical | 10.1 | 573 | 259 | 36 | 9.8 | 137/126 |
| 2 | F | Ia | + | 7 | Metabolic acidosis | Enzyme + clinical + DNA | 4.75 | 1101 | 262 | < 60 | 3.4 | 971/182 |
| 3 | F | Ia | + | 24 | Hepatomegaly | Enzyme + clinical + DNA | 12.5 | 714 | 214 | < 60 | 7.5 | N/A |
| 4 | M | Ia | - | 9 | Hypoglycemia | Histopathology + clinical | N/A | 1646 | 581 | 113 | N/A | 375/348 |
| 5a | F | Ia | + | 1 | Hypoglycemia | Family history + clinical | 7.81 | 719 | N/A | 31 | N/A | N/A |
| 6a | F | Ia | + | 6 | Hypoglycemia | Enzyme + clinical | 7 | 2013 | 226 | 57 | 3 | 142/102 |
| 7 | M | Ia | - | 5 | Hepatomegaly | Enzyme + clinical | 4.6 | 143 | 155 | 116 | 3.4 | 31/40 |
| 8 | M | Ia | + | 4 | Seizure | Enzyme + clinical + DNA | N/A | 216 | 161 | 0 | 6.7 | 48/23 |
| 9 | M | Ia | - | 7 | Hepatomegaly | Enzyme + clinical | 1.7 | 366 | 231 | 77 | 6 | 26/25 |
| 10 | M | Ia | - | 36 | Hypoglycemia | Enzyme + clinical | N/A | 1695 | 285 | 48 | 6.3 | 34 |
| 11 | M | Ia | - | 4 | Hepatomegaly | Clinical + DNA | 4.4 | 836 | 318 | N/A | 8.1 | N/A |
| 12 | F | Ia | - | 48 | Hepatomegaly | Enzyme + clinical | 10.25 | 610 | 185 | 45 | 9 | 59/45 |
| 13 | M | Ia | + | 3 | Hypoglycemia | Enzyme + clinical | N/A | 218 | 187 | 98 | 95/63 | |
| 14 | M | Ia | + | 132 | Short stature | Enzyme + clinical | 8.2 | 941 | 317 | 61 | 10 | 56/69 |
| 15 | M | Ia | - | 7 | Hepatomegaly | Enzyme + clinical + DNA | N/A | 940 | 388 | 32 | 7.3 | 157/130 |
| 16 | F | Ia | - | 84 | Hepatomegaly | Histopathology + clinical | 7.9 | 918 | N/A | 56 | 7.2 | N/A |
| 17 | F | Ia | - | 15 | Hepatomegaly | Histopathology + Clinical | 6 | 355 | 308 | 53 | 5.5 | 33/19 |
| 18 | M | Ib | + | 6 | Hypoglycemia | Enzyme + Clinical | 2.8 | 371 | N/A | 86 | 10.1 | N/A |
| 19 | F | Ib | - | 4 | Recurrent infections | Histopathology + Clinical | 1.6 | 266 | N/A | 143 | 8.9 | N/A |
| 20 | M | Ib | - | 6 | Hypoglycemia | Enzyme + Clinical | N/A | 123 | 107 | N/A | 4.5 | 26/22 |
| 21 | F | Ib | - | 6 | Hypoglycemia | Clinical + DNA | 5.3 | 715 | 196 | 93 | 6.6 | 51/41 |
All patients had clinical manifestations at the time of diagnosis or were in treatment with a pediatric gastroenterologist or medical geneticist at the time of study inclusion and had histopathological evidence of hepatic glycogen buildup and/or G6Pase activity at <10% in liver tissue and/or presence of pathogenic mutations in the G6Pase gene, and absence of high levels of creatine phosphokinase.
AST, aspartate aminotransferase; ALT, alanine aminotransferase.
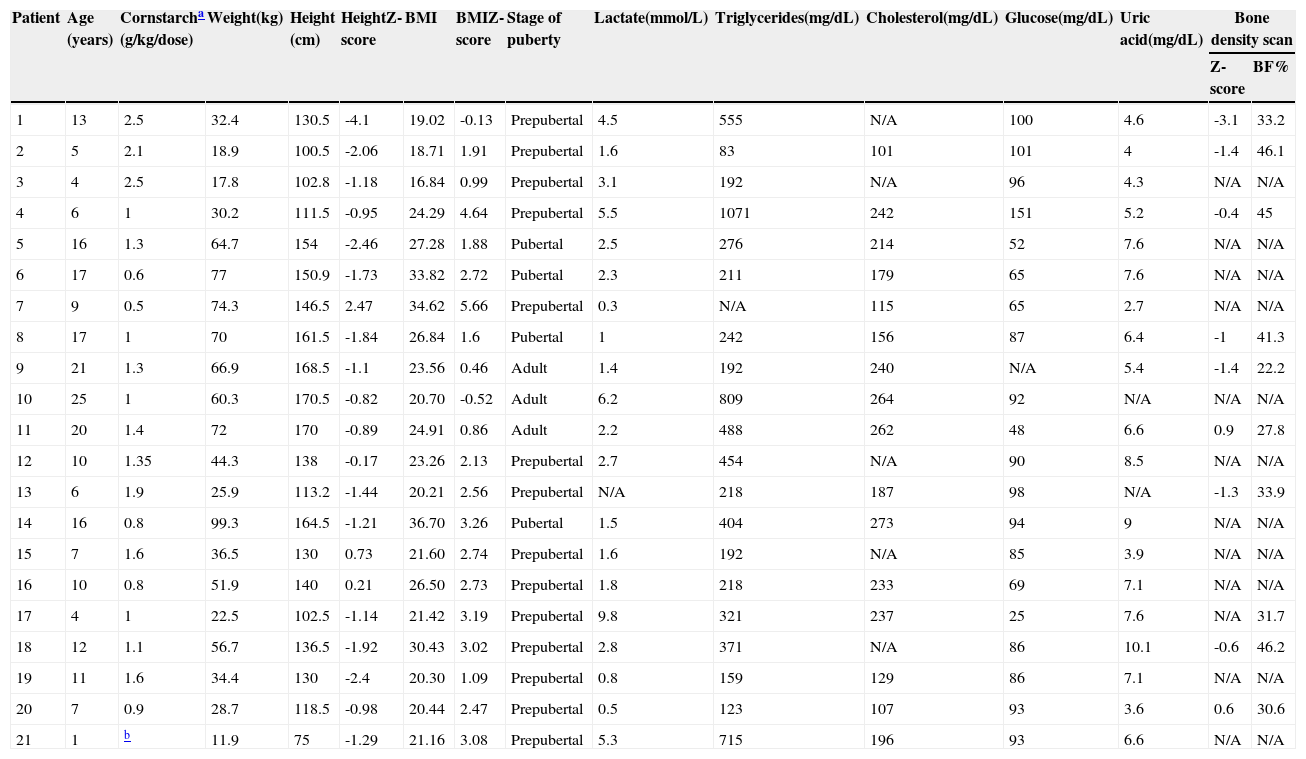
Table 2 presents the anthropometric and laboratory data, as well as compliance with uncooked starch therapy. Sixteen patients had excess body weight (six of 21 severely obese [BMI-for-age zscore >+3]; six of 21 obese; four of 21 overweight). The mean BMI-for-age z-score was 2.19 (1.5 to 2.8), and the mean height-for-age z-score was -1.16 (-1.76 to -0.58); four of 21 patients had short stature, one of whom had very short stature (z-score <-3). Fig. 1 provides a graphical representation of the positive, significant correlation between height and BMI z-scores.
Last anthropometric and laboratory assessment of patients with glycogen storage disorder type I (n=21).
| Patient | Age (years) | Cornstarcha (g/kg/dose) | Weight(kg) | Height (cm) | HeightZ-score | BMI | BMIZ-score | Stage of puberty | Lactate(mmol/L) | Triglycerides(mg/dL) | Cholesterol(mg/dL) | Glucose(mg/dL) | Uric acid(mg/dL) | Bone density scan | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z-score | BF% | ||||||||||||||
| 1 | 13 | 2.5 | 32.4 | 130.5 | -4.1 | 19.02 | -0.13 | Prepubertal | 4.5 | 555 | N/A | 100 | 4.6 | -3.1 | 33.2 |
| 2 | 5 | 2.1 | 18.9 | 100.5 | -2.06 | 18.71 | 1.91 | Prepubertal | 1.6 | 83 | 101 | 101 | 4 | -1.4 | 46.1 |
| 3 | 4 | 2.5 | 17.8 | 102.8 | -1.18 | 16.84 | 0.99 | Prepubertal | 3.1 | 192 | N/A | 96 | 4.3 | N/A | N/A |
| 4 | 6 | 1 | 30.2 | 111.5 | -0.95 | 24.29 | 4.64 | Prepubertal | 5.5 | 1071 | 242 | 151 | 5.2 | -0.4 | 45 |
| 5 | 16 | 1.3 | 64.7 | 154 | -2.46 | 27.28 | 1.88 | Pubertal | 2.5 | 276 | 214 | 52 | 7.6 | N/A | N/A |
| 6 | 17 | 0.6 | 77 | 150.9 | -1.73 | 33.82 | 2.72 | Pubertal | 2.3 | 211 | 179 | 65 | 7.6 | N/A | N/A |
| 7 | 9 | 0.5 | 74.3 | 146.5 | 2.47 | 34.62 | 5.66 | Prepubertal | 0.3 | N/A | 115 | 65 | 2.7 | N/A | N/A |
| 8 | 17 | 1 | 70 | 161.5 | -1.84 | 26.84 | 1.6 | Pubertal | 1 | 242 | 156 | 87 | 6.4 | -1 | 41.3 |
| 9 | 21 | 1.3 | 66.9 | 168.5 | -1.1 | 23.56 | 0.46 | Adult | 1.4 | 192 | 240 | N/A | 5.4 | -1.4 | 22.2 |
| 10 | 25 | 1 | 60.3 | 170.5 | -0.82 | 20.70 | -0.52 | Adult | 6.2 | 809 | 264 | 92 | N/A | N/A | N/A |
| 11 | 20 | 1.4 | 72 | 170 | -0.89 | 24.91 | 0.86 | Adult | 2.2 | 488 | 262 | 48 | 6.6 | 0.9 | 27.8 |
| 12 | 10 | 1.35 | 44.3 | 138 | -0.17 | 23.26 | 2.13 | Prepubertal | 2.7 | 454 | N/A | 90 | 8.5 | N/A | N/A |
| 13 | 6 | 1.9 | 25.9 | 113.2 | -1.44 | 20.21 | 2.56 | Prepubertal | N/A | 218 | 187 | 98 | N/A | -1.3 | 33.9 |
| 14 | 16 | 0.8 | 99.3 | 164.5 | -1.21 | 36.70 | 3.26 | Pubertal | 1.5 | 404 | 273 | 94 | 9 | N/A | N/A |
| 15 | 7 | 1.6 | 36.5 | 130 | 0.73 | 21.60 | 2.74 | Prepubertal | 1.6 | 192 | N/A | 85 | 3.9 | N/A | N/A |
| 16 | 10 | 0.8 | 51.9 | 140 | 0.21 | 26.50 | 2.73 | Prepubertal | 1.8 | 218 | 233 | 69 | 7.1 | N/A | N/A |
| 17 | 4 | 1 | 22.5 | 102.5 | -1.14 | 21.42 | 3.19 | Prepubertal | 9.8 | 321 | 237 | 25 | 7.6 | N/A | 31.7 |
| 18 | 12 | 1.1 | 56.7 | 136.5 | -1.92 | 30.43 | 3.02 | Prepubertal | 2.8 | 371 | N/A | 86 | 10.1 | -0.6 | 46.2 |
| 19 | 11 | 1.6 | 34.4 | 130 | -2.4 | 20.30 | 1.09 | Prepubertal | 0.8 | 159 | 129 | 86 | 7.1 | N/A | N/A |
| 20 | 7 | 0.9 | 28.7 | 118.5 | -0.98 | 20.44 | 2.47 | Prepubertal | 0.5 | 123 | 107 | 93 | 3.6 | 0.6 | 30.6 |
| 21 | 1 | b | 11.9 | 75 | -1.29 | 21.16 | 3.08 | Prepubertal | 5.3 | 715 | 196 | 93 | 6.6 | N/A | N/A |
BMI, body mass index.
All patients were on uncooked starch therapy (four to six times/day) and none were on continuous nocturnal glucose infusion.
Patient who received cornstarch irregularly.
Height-for-age and BMI-for-age Z-scores calculated in World Health Organization's Anthro and AnthroPlus. Bone density scanning performed in a Lunar iDXA (GE Healthcare) device. Bone mineral density Z-scores calculated and body composition expressed as body fat percentage (BF%). Bone mineral density Z-scores not calculated for patient 17 due to age < 5 years. N/A, not available.
Reference ranges: lactate, 0.5–2.2 mmol/L; triglycerides, ≤100mg/dL at age < 10 years, ≤130mg/dL at age 10–19, ≤150mg/dL in adults; total cholesterol, < 129mg/dL; glucose, 60–99mg/dL; uric acid, 2.4–7mg/dL.
. BMI, body mass index.")
Body composition was analyzed in ten patients (eight with GSDIa, two with GSDIb) (Table 2). Fourteen patients underwent abdominal ultrasonography for assessment of liver size; of these, five had a normal liver size, one of whom had a visible hepatic nodule. The eight remaining patients had hepatomegaly, and two had more than three detectable nodules.
DiscussionThe characterization of the natural history of rare diseases and of the efficacy of treatments for these conditions is always hindered by small sample sizes.10 The small number of patients is attributable not only to the rarity of these diseases, but also to underdiagnosis, particularly of cases with relatively mild clinical manifestations. Therefore, studies such as the present – the first-ever characterization of a population of GSDI patients in Brazil are paramount, given their crucial role in enabling later conduction of meta-analysis and the drawing of more robust conclusions.
Diagnosis of GSDI was delayed in this sample, confirming the initial hypothesis. According to the literature, the usual age of symptom onset in patients with GSDI is 3 months.4 This study did not assess the variable “age at symptom onset” as the authors believe it to be subject to a wide range of biases, particularly recall bias. Studies have shown that earlier diagnosis and treatment onset are associated with lower odds of complications.3 In the present sample, the earliest clinical diagnosis was established in a patient (patient 5) who developed symptoms before the 1st month of life, who had an older sister (patient 6) with a confirmed diagnosis of GSDIa. The latest diagnosis was at 132 months of age, in patient 14, who had subclinical hypoglycemia and was diagnosed after a three year investigation prompted by short stature, thus representing a somewhat attenuated phenotype of the condition. Although hypoglycemia is one of the cardinal symptoms that drive clinical suspicion of GSDI, it may sometimes go unnoticed due to use of lactic acid as a substrate for cerebral metabolism.11 Therefore, even though symptomatic hypoglycemia is frequently reported, its absence does not rule out a diagnosis of GSDI. In 2003, Shieh et al. published a case report describing delayed diagnosis of GSDI and suggested that “milder” forms of the condition may occur.11 Also in 2003, an article recommended that adolescents with unexplained hyperuricemia and hyperlipidemia should be screened for GSDI, even if hypoglycemia was absent.12
Regarding diagnostic procedures, most patients in the present sample underwent a liver biopsy.13 This finding is rather surprising in view of the increased worldwide accessibility of genetic testing. The G6PC gene is small (12.5kb, 5 exons) and thus easily sequenced; furthermore, the variants p.347X and p.R83C appear to be common in the Brazilian population, as reported by Reis et al. in 2011.14 In the present sample, these mutations were found in four of ten and three of ten patients with GSDIa, respectively (data not shown). Although it is not entirely devoid of risk, blood collection for genetic assays is a far less invasive and less costly procedure than liver biopsy for histopathological examination or enzyme activity assessment. Isolated histological analysis of liver tissue without measurement of enzyme activity is not sufficient to determine the type of GSD, although it can demonstrate glycogen and fat deposition, and is valuable in the differential diagnosis of other liver diseases. Conversely, enzyme assays are available only at very few centers and are associated with a series of logistical challenges, such as tissue transport (specimens should preferably be fresh or frozen) to the reference laboratory.
The present data suggest a trend toward patients with higher height-for-age Z-scores having higher BMI-for-age Z-scores as well.15 Although this trend was affected by outliers, it suggests that intensive dietary management leads to better growth at the expense of marked weight gain, as previously reported by Weinstein and Wolfsdorf in 2002.6 Management of obesity in patients with GSDI is certainly a topic deserving of greater research attention.
Growth retardation is a finding of major importance in children with GSDI,16 and short stature is common in adults with the condition. In the present sample, patients with inadequate metabolic control according to the ESGSD I4 had the worst height-for-age Z-scores. The pathophysiology of short stature in GSDI has yet to be elucidated, but studies conducted since 2008 have shown that good metabolic control can improve growth.17,18 Hormonal changes, variation in blood pH (due to metabolic acidosis), and hyperlactatemia may contribute to this growth deficit. According to the ESGSD I criteria,5 half of all patients in the present sample had good metabolic control of their condition, even though some had BMI Z-scores > 2 SDs, which may account at least in part for the near-adequate growth of this population (18 of 21 patients had height-for-age Z-scores > -2 SDs).
The purpose of dietary management of GSDI is to mimic endogenous glucose production. Exogenous dextrose administration strategies for maintenance of normoglycemia have been assessed and modified in recent years. Frequent meals containing partially cooked starch, continuous nocturnal gastric drip feeding (CNGDF) of dextrose via nasogastric tube, and uncooked cornstarch (UCCS) therapy are some of the available strategies. None of the patients in this case series were on continuous nocturnal feeding; all were on uncooked cornstarch therapy (five to six doses per 24h, including overnight). A recent meta-analysis15 compared several studies of UCCS (diurnal and nocturnal) with studies of CNGDF, and found both short-term and long-term improvement of metabolic control in patients given UCCS.19 Therefore, CNGDF should be restricted to select cases, as the inconvenience of use of a feeding pump and the risk of severe hypoglycemia in case of abrupt discontinuation of feeding (e.g., due to a power outage or pump malfunction) do not outweigh the metabolic control benefits of intermittent night-time UCCS administration. A modified cornstarch formulation (Glycosade®, Vitaflo, Nestlé Health Nutrition, Vevey, Switzerland), which granted the Food and Drug Administration (FDA) approval in 2012, is an alternative that may allow patients to sleep through the night.20
The hepatomegaly observed in patients with GSDI may be the result of glycogen deposition and fatty liver disease secondary to increased flow of free fatty acids from the adipose tissue to the liver.21 Routine liver ultrasound is a noninvasive diagnostic modality that can be used to assess long-term treatment success. As this study's analysis of ultrasound findings was based on a chart review, operator variability is a concern. The histology of hepatocellular adenomas in GSDI is similar to that of adenomas seen in other conditions. Several hypotheses have attempted to explain the development of adenomatous changes, such as imbalances in glucagon-to-insulin ratio, cellular glycogen overload, and proto-oncogene activation.21 The three patients with hepatic adenomas in the present case series were 16, 17, and 25 years old (patients 5, 6, and 10 respectively). Patients 5 and 6 had poor metabolic control, with hyperuricemia and hypoglycemia despite low triglycerides and near-normal lactate levels. Patient 10, the oldest patient in this sample, also had inadequate metabolic control.
Hepatocellular adenomas may occur in 22% to 75% of adults with GSDIa, and the risk of malignant transformation is approximately 10%.21 As most patients are under the age of 20, a low incidence of adenomas is to be expected in this population regardless of metabolic control. Thus far, there are no cases of hepatocarcinoma in this series.
Of the nine patients in whom bone density scans were performed, only one (patient 1) had low bone mass for chronological age in accordance with the 2008 Official Position Statement of the Brazilian Society for Bone Densitometry.22 Several mechanisms have been suggested to explain the low bone mineral density observed in patients with GSDI: persistent acidosis, urinary calcium loss without adequate replacement, reduction of bone matrix (hypoglycemia leads to decreased glycosylation of bone matrix proteins), and changes in growth hormone (GH) levels.18 Furthermore, many patients with GSDI exhibit abnormal pubertal growth,5 and sex steroids play a major role in bone formation, particularly during puberty.22 Some issues must be taken into account when analyzing bone mineral density in children and adolescents, such as bone maturation, sex, and stage of puberty.23 Overall, the patients assessed in this case series had good bone mineral density despite their GSDI.
Despite the rarity of GSDI, it must not be overlooked by pediatricians. The agent used for treatment of this inborn error of metabolism is readily available at any grocery store or supermarket. Administration of cornstarch as a nutraceutical (food used for medicinal purposes) to GSDI patients leads to excessive weight gain as a side effect, due to the attendant increase in total carbohydrate intake and probably due to relative physical inactivity as well. No studies have assessed the efficacy of physical exercise in patients with GSDI, and it is not contraindicated. Therefore, physical activity – supported by a well-designed dietary prescription that takes the pre- and post-exercise periods into account – may be an effective strategy for the control of weight gain in patients whose metabolic control is otherwise satisfactory.
Greater awareness of this disorder among pediatricians should aid their search for an etiological diagnosis in cases of hypoglycemia, hepatomegaly, dyslipidemia, and short stature that might otherwise be improperly managed. Early diagnosis based on clinical and laboratory findings is feasible, easy, and affordable even where access to specialty care is limited. Nevertheless, investment in centers for expert molecular diagnosis is both warranted and necessary, as the use of molecular methods practically obviates the need for liver biopsy. Early treatment can be instituted at any health service, does not require any complex interventions, and decreases the risk of death, mainly by preventing severe hypoglycemia. Adequately treated patients can lead intellectually and socially satisfying lives with no limitations other than a special diet.
FundingFIPE-HCPA, FAPERGS and CNPq.
Conflicts of interestThe authors declare no conflicts of interest.
This work was supported by FIPE-HCPA, FAPERGS and CNPq. The authors would like to thank the multidisciplinary team at the Inborn Errors of Metabolism Clinics of the HCPA Medical Genetics Service, the HCPA and PUC Gastroenterology and Hepatology Services, the SIEM, and Ana Carolina Monteiro for helping with the manuscript and with the diagnosis and assistance of the patients. The authors would also like to thank Dr. Terry Derks for the invaluable learning opportunities provided, and Dr. David Weinstein for his many teachings that contributed directly or indirectly to this study, which may yet become the driving force for a multicenter research group.
Please cite this article as: Santos BL, de Souza CF, Schuler-Faccini L, Refosco L, Epifanio M, Nalin T, et al. Glycogen storage disease type I: clinical and laboratory profile. J Pediatr (Rio J). 2014;90:572–79.
Study conducted at the Universidade Federal do Rio Grande do Sul, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brazil.




