

To characterize the clinical, laboratory, and anthropometric profile of a sample of Brazilian patients with glycogen storage disease type I managed at an outpatient referral clinic for inborn errors of metabolism.
MethodsThis was a cross‐sectional outpatient study based on a convenience sampling strategy. Data on diagnosis, management, anthropometric parameters, and follow‐up were assessed.
ResultsTwenty‐one patients were included (median age 10 years, range 1–25 years), all using uncooked cornstarch therapy. Median age at diagnosis was 7 months (range, 1–132 months), and 19 patients underwent liver biopsy for diagnostic confirmation. Overweight, short stature, hepatomegaly, and liver nodules were present in 16 of 21, four of 21, nine of 14, and three of 14 patients, respectively. A correlation was found between height‐for‐age and BMI‐for‐age Z‐scores (r=0.561; p=0.008).
ConclusionsDiagnosis of glycogen storage disease type I is delayed in Brazil. Most patients undergo liver biopsy for diagnostic confirmation, even though the combination of a characteristic clinical presentation and molecular methods can provide a definitive diagnosis in a less invasive manner. Obesity is a side effect of cornstarch therapy, and appears to be associated with growth in these patients.
Caracterizar o perfil clínico, laboratorial e antropométrico de uma amostra de pacientes brasileiros com doença de depósito de glicogênio tipo I tratados em um ambulatório de referência para erros inatos do metabolismo.
MétodosEste foi um estudo ambulatorial transversal com base em uma estratégia de amostragem de conveniência. Foram avaliados os dados com relação ao diagnóstico, tratamento, parâmetros antropométricos e acompanhamento.
ResultadosForam incluídos 21 pacientes (idade média de 10 anos, faixa 1‐25 anos de idade), e todos se encontravam em terapia de amido de milho cru. A idade média na época do diagnóstico foi de sete meses (faixa, 1‐32 meses), e 19 pacientes foram submetidos a biópsia hepática para confirmação do diagnóstico. Sobrepeso, baixa estatura, hepatomegalia e nódulos hepáticos foram fatores presentes em 16 de 21, quatro de 21, nove de 14 e três de 14 pacientes, respectivamente. Foi encontrada uma correlação entre os escores z para peso para idade e IMC para idade (r=0,561; p=0,008).
ConclusõesO diagnóstico da doença de depósito de glicogênio tipo I tem sido tardio no Brasil. A maioria dos pacientes foi submetida a confirmação do diagnóstico, apesar de o quadro clínico característico e os métodos moleculares poderem fornecer um diagnóstico definitivo de forma menos invasiva. Obesidade é um efeito colateral da terapia com amido de milho e parece estar associada a crescimento nesses pacientes.
A doença de depósito de glicogênio tipo I (GDSI, doença de von Gierke) é causada pela deficiência de glicose‐6‐fosfatase (G6Pase), uma enzima que catalisa a hidrólise de glicose‐6‐fosfato (G6P) em glicose e fosfato inorgânico (Pi), um passo importante na manutenção da homeostase da glicose. São reconhecidos dois subtipos principais da GSDI: GSD tipo Ia (GSDIa), que é o resultado de uma mutação que afeta a subunidade catalítica da G6Pase‐alfa (ou G6PC), e GSD tipo Ib (GSDIb), que é causada por um defeito na translocase de G6P (ou G6PT).1 A GSDI é herdada em um padrão autossômico recessivo e sua incidência é estimada em um para 100.000 nascidos vivos, sendo a GSD hepática mais comum.2
Pacientes com GSDIa apresentam hepatomegalia, uma característica de “fácies de boneca”, baixa estatura e fadiga crônica. Os achados laboratoriais sugestivos de GSDIa incluem hipoglicemia após quatro a seis horas de jejum, acidose láctica, hipertrigliceridemia e hiperuricemia. Os testes funcionais para um diagnóstico diferenciado de hipoglicemia mostram ausência de resposta glicêmica a injeção de glucagon e piora da hiperlactatemia,3 ao passo que a análise histopatológica da biópsia hepática mostra acúmulo de glicogênio no fígado. Na GSD tipo Ib, o quadro clínico é bem semelhante ao da GSDIa, porém pode ser acompanhada de neutropenia, com infecções recorrentes (principalmente do trato gastrointestinal) e uma maior incidência de doença inflamatória do intestino.4 Apesar de os métodos padrão‐ouro para diagnóstico da GSDIa serem avaliação da atividade de G6PC ou G6PT no tecido hepático e/ou detecção de mutações patogênicas nos genes que codificam a G6PC e G6PT, pode se dar início a uma terapia específica com base exclusivamente nos achados clínicos e histopatológicos.3 O acesso aos testes de DNA/enzimáticos é limitado, pois eles são disponibilizados apenas a poucos centros nacionais e internacionais selecionados, normalmente no âmbito de projetos de pesquisa.
O tratamento da GSDI é basicamente alimentar3 e consiste em refeições frequentes – preferencialmente que contenham carboidratos de liberação lenta, como amido de milho cru – em intervalos regulares e com restrição quanto à ingestão de frutose, sacarose e lactose. Em neonatos, a estratégia de tratamento recomendada inclui alimentações frequentes e infusão contínua de glicose à noite a uma taxa de 6‐8mg/kg/min por meio de uma sonda nasogástrica ou de gastrostomia. A eficácia do tratamento é medida pelo monitoramento dos parâmetros de crescimento e bioquímicos, bem como por ultrassom abdominal para avaliar o volume do fígado e a presença de nódulos. O manejo dietético adequado reduz o risco de complicações de longo prazo, que incluem baixa estatura, osteoporose ou perda mineral óssea, doença renal com hipertensão, proteinúria, cálculos renais, nefrocalcinose, adenomas hepatocelulares (com potencial de transformação maligna), pancreatite secundária a hipertrigliceridemia e hipoglicemia potencialmente fatal.5,6
O objetivo deste estudo foi avaliar o perfil clínico e laboratorial de uma amostra de pacientes brasileiros com GSDI recrutados de um ambulatório de referência para erros inatos do metabolismo. Nossa principal hipótese de pesquisa foi que o diagnóstico da GSDI tem sido tardio no Brasil, tanto por falta de acesso a métodos de diagnóstico quanto por má conscientização da condição por prestadores de serviços de saúde, dificultando, assim, um rápido acesso a tratamento e aconselhamento genético.
MétodosEste estudo foi aprovado pelo Comitê de Ética do Hospital de Clínicas de Porto Alegre (HCPA, Brasil). Foi obtido consentimento informado por escrito de todos os indivíduos antes da participação no estudo.
Esta foi uma casuística ambulatorial com análise transversal das variáveis de interesse. Foi utilizada uma estratégia de amostragem de conveniência. O estudo foi realizado entre março de 2011 e janeiro de 2013. O critério de inclusão foi um diagnóstico de GSDI estabelecido utilizando pelo menos dois dos seguintes métodos (o diagnóstico foi confirmado em todos pacientes de forma independente pelos autores): a) Diagnóstico Clínico, definido por mais de 12 meses de cuidados especializados (acompanhado por um hepatologista ou médico geneticista) e manifestações clínicas compatíveis com GSDI (hipoglicemia com hiperlactatemia, hipertrigliceridemia, hiperuricemia, hepatomegalia e/ou déficit de crescimento e baixa estatura e níveis normais de creatinofosfoquinase [CPK]) no momento do diagnóstico ou da inclusão no estudo; b) Histórico Familiar Positivo compatível com herança autossômica recessiva, desde que a GSDI tenha sido confirmada por método enzimáticos ou análise de DNA no(s) parente(s) afetado(s); c) Diagnóstico Histopatológico, definido como presença de alterações histológicas no tecido hepático compatíveis com GSD, como núcleos hiperglicogenados, fibrose discreta e alterações de gordura com vacúolos lipídicos;7 d) Diagnóstico Enzimático, definido por atividade negligenciável (< 10%) da G6Pase em amostras de tecido hepático frescas ou congeladas; ou e) Diagnóstico Molecular, definido pela presença de mutações patogênicas no gene G6PC (para pacientes com GSDIa) ou no gene SLC37A3 (para os com GSDIb), detectadas por métodos moleculares. Essa distinção entre GSDIa e GSDIb foi principalmente com base em achados clínicos (ausência ou presença de neutropenia, respectivamente), pois os diagnósticos moleculares não estavam disponíveis para a maioria dos pacientes.
Os pacientes foram convidados a participar do estudo após consultas de rotina. Os que concordaram em participar foram todos avaliados pelo mesmo pesquisador e foram submetidos a verificação direcionada do histórico, exame físico e avaliação antropométrica. Os últimos valores laboratoriais (glicemia, lactato, colesterol, triglicerídeos, ácido úrico) e achados de imagem disponíveis para cada paciente foram obtidos por meio de uma revisão de prontuários. Os exames realizados até três meses antes da avaliação antropométrica foram considerados aceitáveis. As variáveis de interesse foram sexo, consanguinidade, idade atual, idade na época do diagnóstico (definido como idade na qual os pais relataram um diagnóstico específico de GSD ou, se indisponível, a idade na época do diagnóstico conforme observado no primeiro prontuário do paciente contendo os resultados dos testes de diagnóstico e o início do manejo dietético), parâmetros laboratoriais (atuais e no momento do diagnóstico), biópsia hepática para exame histopatológico ou análise molecular, dados clínicos e de imagem atuais (avaliação antropométrica, ultrassom do fígado, densidade mineral óssea e composição corporal por meio de absorciometria de dupla energia de raios‐X [DEXA]).
A avaliação antropométrica consistiu da avaliação de peso (kg) e altura (cm). O peso corporal foi avaliado utilizando escalas digitais com uma capacidade máxima de 150kg e uma resolução de 100g, certificados pelo Instituto Nacional de Metrologia, Qualidade e Tecnologia (Inmetro). Os pacientes foram pesados nus e descalços. O peso foi medido com um estadiômetro de parede com precisão de 1mm. Em adolescentes, foi utilizada a escala de Tanner. As medidas e classificações antropométricas para idade e sexo foram calculadas no conjunto de software AnthroPlus da OMS. As variáveis de interesse foram escores z para peso e IMC para idade, conforme proposto pela Sociedade Brasileira de Pediatria.8
O tamanho do fígado foi medido por ultrassonografia e avaliado para normalidade com base nos tamanhos de referência para crianças publicados em 2010 por Dhingra et al.9 Quando não disponíveis os dados objetivos sobre o tamanho do fígado, utilizamos, em vez disso, a impressão do ultrassonografista (normal ou aumentado).
Os critérios de adequação do controle metabólico tiveram com base o Estudo Europeu sobre Doença de Depósito de Glicogênio Tipo I (ESGSD I):5 glicemia > 63mg/dL, triglicerídeos < 530mg/dL, ácido úrico < 7mg/dL, IMC entre desvios‐padrão 0 e +2 e lactato >2,5 mmol/L (este utilizado como relação lactato urinário/creatinina não estava disponível). A ausência de adenomas hepáticos e altura para peso adequada (escore z > ‐2SD) são parâmetros importantes para a avaliação da adequação do controle metabólico, porém não fazem parte do ESGSD I.5
Foram realizadas análises estatísticas no ambiente de software 20.0 do Pacote Estatístico para as Ciências Sociais® (SPSS Inc., Chicago, IL, EUA). As variáveis contínuas foram expressas como médias e desvios‐padrão ou medianas e intervalos interquartis. A análise da variância (ANOVA) foi utilizada para comparação de escores z para altura e IMC. O nível de significância foi estabelecido em 5%. Os dados foram inseridos em uma planilha do Microsoft Excel 2010 para Windows (Microsoft, Redmond, WA, USA) e analisados no SPSS 20.0 (IBM Corp., Armonk, NY, EUA).
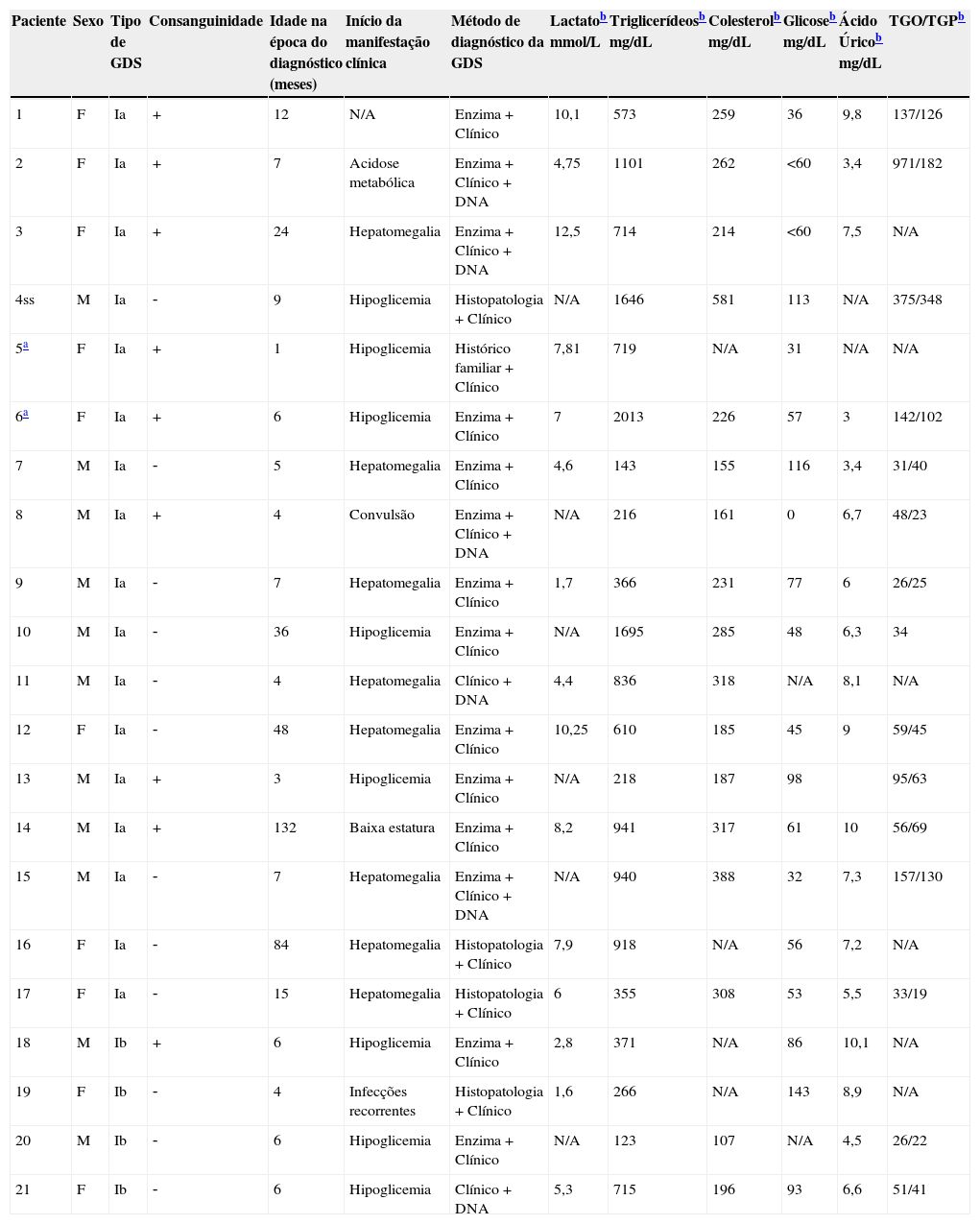
ResultadosNo total, 21 pacientes foram incluídos no estudo: 17 com GSDIa e quatro com GSDIb. A tabela 1 apresenta o perfil da amostra no momento do diagnóstico.
Resumo dos achados no diagnóstico dentre pacientes com doença de depósito de glicogênio (GDS) tipo I (n=21)
| Paciente | Sexo | Tipo de GDS | Consanguinidade | Idade na época do diagnóstico (meses) | Início da manifestação clínica | Método de diagnóstico da GDS | Lactatob mmol/L | Triglicerídeosb mg/dL | Colesterolb mg/dL | Glicoseb mg/dL | Ácido Úricob mg/dL | TGO/TGPb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Ia | + | 12 | N/A | Enzima + Clínico | 10,1 | 573 | 259 | 36 | 9,8 | 137/126 |
| 2 | F | Ia | + | 7 | Acidose metabólica | Enzima + Clínico + DNA | 4,75 | 1101 | 262 | <60 | 3,4 | 971/182 |
| 3 | F | Ia | + | 24 | Hepatomegalia | Enzima + Clínico + DNA | 12,5 | 714 | 214 | <60 | 7,5 | N/A |
| 4ss | M | Ia | ‐ | 9 | Hipoglicemia | Histopatologia + Clínico | N/A | 1646 | 581 | 113 | N/A | 375/348 |
| 5a | F | Ia | + | 1 | Hipoglicemia | Histórico familiar + Clínico | 7,81 | 719 | N/A | 31 | N/A | N/A |
| 6a | F | Ia | + | 6 | Hipoglicemia | Enzima + Clínico | 7 | 2013 | 226 | 57 | 3 | 142/102 |
| 7 | M | Ia | ‐ | 5 | Hepatomegalia | Enzima + Clínico | 4,6 | 143 | 155 | 116 | 3,4 | 31/40 |
| 8 | M | Ia | + | 4 | Convulsão | Enzima + Clínico + DNA | N/A | 216 | 161 | 0 | 6,7 | 48/23 |
| 9 | M | Ia | ‐ | 7 | Hepatomegalia | Enzima + Clínico | 1,7 | 366 | 231 | 77 | 6 | 26/25 |
| 10 | M | Ia | ‐ | 36 | Hipoglicemia | Enzima + Clínico | N/A | 1695 | 285 | 48 | 6,3 | 34 |
| 11 | M | Ia | ‐ | 4 | Hepatomegalia | Clínico + DNA | 4,4 | 836 | 318 | N/A | 8,1 | N/A |
| 12 | F | Ia | ‐ | 48 | Hepatomegalia | Enzima + Clínico | 10,25 | 610 | 185 | 45 | 9 | 59/45 |
| 13 | M | Ia | + | 3 | Hipoglicemia | Enzima + Clínico | N/A | 218 | 187 | 98 | 95/63 | |
| 14 | M | Ia | + | 132 | Baixa estatura | Enzima + Clínico | 8,2 | 941 | 317 | 61 | 10 | 56/69 |
| 15 | M | Ia | ‐ | 7 | Hepatomegalia | Enzima + Clínico + DNA | N/A | 940 | 388 | 32 | 7,3 | 157/130 |
| 16 | F | Ia | ‐ | 84 | Hepatomegalia | Histopatologia + Clínico | 7,9 | 918 | N/A | 56 | 7,2 | N/A |
| 17 | F | Ia | ‐ | 15 | Hepatomegalia | Histopatologia + Clínico | 6 | 355 | 308 | 53 | 5,5 | 33/19 |
| 18 | M | Ib | + | 6 | Hipoglicemia | Enzima + Clínico | 2,8 | 371 | N/A | 86 | 10,1 | N/A |
| 19 | F | Ib | ‐ | 4 | Infecções recorrentes | Histopatologia + Clínico | 1,6 | 266 | N/A | 143 | 8,9 | N/A |
| 20 | M | Ib | ‐ | 6 | Hipoglicemia | Enzima + Clínico | N/A | 123 | 107 | N/A | 4,5 | 26/22 |
| 21 | F | Ib | ‐ | 6 | Hipoglicemia | Clínico + DNA | 5,3 | 715 | 196 | 93 | 6,6 | 51/41 |
Todos os pacientes apresentaram manifestações clínicas na época do diagnóstico ou estavam em tratamento com um gastroenterologista pediátrico ou médico geneticista na época da inclusão no estudo e apresentaram evidência histopatológica de acúmulo de glicogênio hepático e/ou atividade da G6Pase a < 10% no tecido hepático e/ou presença de mutações patogênicas no gene da G6Pase e ausência de altos níveis de creatinofosfoquinase.
Valores na época do diagnóstico; +, presença; ‐, ausência; N/A, não aplicável. Intervalos de referência: lactato, 0,5–2,2 mmol/L; triglicerídeos, ≤ 100mg/dL em idades < 10 anos, ≤ 130mg/dL em idades entre 10‐19 anos, ≤ 150mg/dL em adultos; colesterol total, < 129mg/dL; glicose, 60‐99mg/dL; ácido úrico, 2,4–7mg/dL; TGO < 37mg/dL; TGP, < 41mg/dL.
TGO, transaminase glutâmico‐oxalacética; TGP, transaminase glutâmico pirúvica.
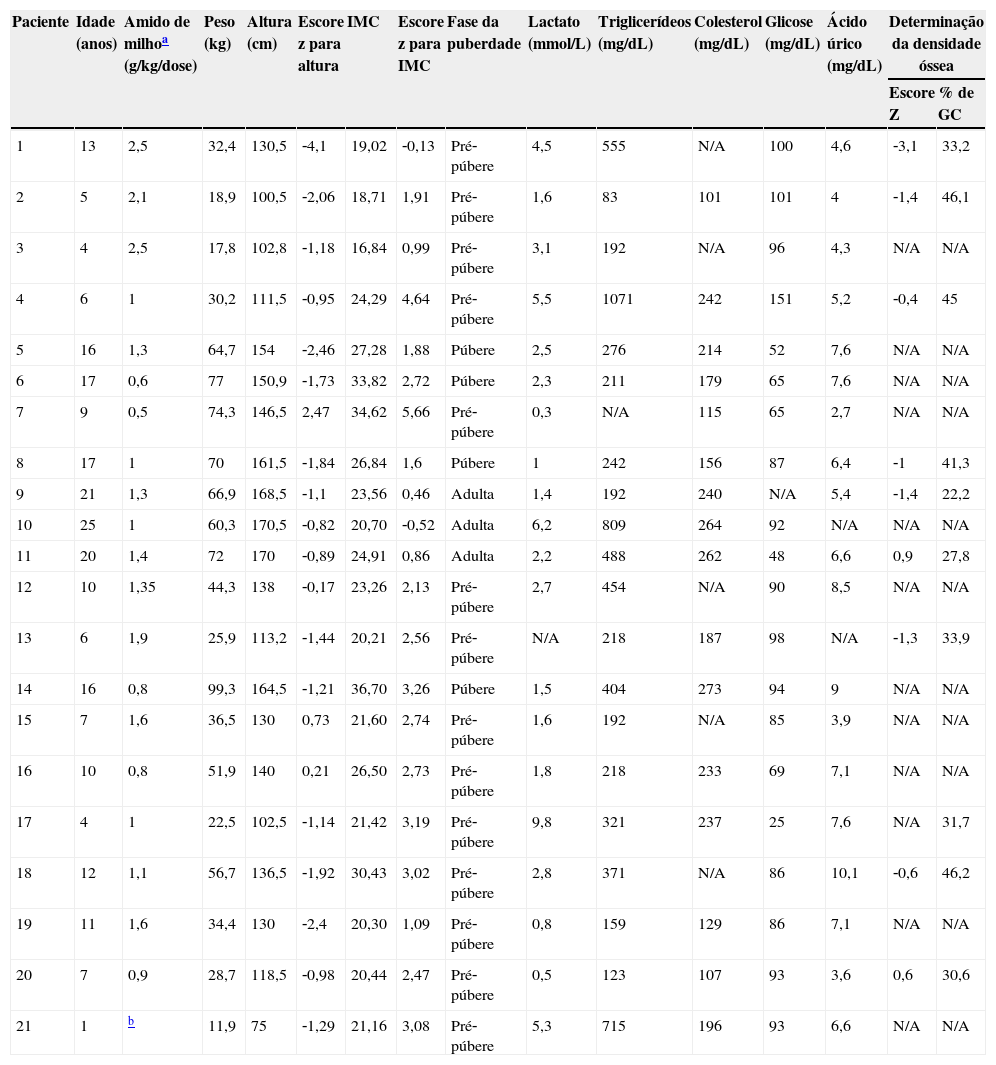
A tabela 2 mostra os dados antropométricos e laboratoriais, bem como a conformidade com a terapia de amido de milho cru. Dentre os pacientes, 16 apresentaram excesso de peso corporal (seis de 21 obesidade grave [escore z para IMC para idade > +3]; seis de 21 obesidade; quatro de 21 sobrepeso). O escore z médio para IMC para idade foi de 2,19 (1,5 a 2,8) e o escore z para altura para idade foi de ‐1,16 (‐1,76 a ‐0,58); quatro de 21 pacientes apresentaram baixa estatura, um dos quais apresentou muito baixa estatura (escore z < ‐3). A figura 1 mostra uma representação gráfica da correlação positiva e significativa entre os escores z para altura e IMC.
Última avaliação antropométrica e laboratorial de pacientes com doença de depósito de glicogênio tipo I (n=21)
| Paciente | Idade (anos) | Amido de milhoa (g/kg/dose) | Peso (kg) | Altura (cm) | Escore z para altura | IMC | Escore z para IMC | Fase da puberdade | Lactato (mmol/L) | Triglicerídeos (mg/dL) | Colesterol (mg/dL) | Glicose (mg/dL) | Ácido úrico (mg/dL) | Determinação da densidade óssea | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Escore Z | % de GC | ||||||||||||||
| 1 | 13 | 2,5 | 32,4 | 130,5 | ‐4,1 | 19,02 | ‐0,13 | Pré‐púbere | 4,5 | 555 | N/A | 100 | 4,6 | ‐3,1 | 33,2 |
| 2 | 5 | 2,1 | 18,9 | 100,5 | ‐2,06 | 18,71 | 1,91 | Pré‐púbere | 1,6 | 83 | 101 | 101 | 4 | ‐1,4 | 46,1 |
| 3 | 4 | 2,5 | 17,8 | 102,8 | ‐1,18 | 16,84 | 0,99 | Pré‐púbere | 3,1 | 192 | N/A | 96 | 4,3 | N/A | N/A |
| 4 | 6 | 1 | 30,2 | 111,5 | ‐0,95 | 24,29 | 4,64 | Pré‐púbere | 5,5 | 1071 | 242 | 151 | 5,2 | ‐0,4 | 45 |
| 5 | 16 | 1,3 | 64,7 | 154 | ‐2,46 | 27,28 | 1,88 | Púbere | 2,5 | 276 | 214 | 52 | 7,6 | N/A | N/A |
| 6 | 17 | 0,6 | 77 | 150,9 | ‐1,73 | 33,82 | 2,72 | Púbere | 2,3 | 211 | 179 | 65 | 7,6 | N/A | N/A |
| 7 | 9 | 0,5 | 74,3 | 146,5 | 2,47 | 34,62 | 5,66 | Pré‐púbere | 0,3 | N/A | 115 | 65 | 2,7 | N/A | N/A |
| 8 | 17 | 1 | 70 | 161,5 | ‐1,84 | 26,84 | 1,6 | Púbere | 1 | 242 | 156 | 87 | 6,4 | ‐1 | 41,3 |
| 9 | 21 | 1,3 | 66,9 | 168,5 | ‐1,1 | 23,56 | 0,46 | Adulta | 1,4 | 192 | 240 | N/A | 5,4 | ‐1,4 | 22,2 |
| 10 | 25 | 1 | 60,3 | 170,5 | ‐0,82 | 20,70 | ‐0,52 | Adulta | 6,2 | 809 | 264 | 92 | N/A | N/A | N/A |
| 11 | 20 | 1,4 | 72 | 170 | ‐0,89 | 24,91 | 0,86 | Adulta | 2,2 | 488 | 262 | 48 | 6,6 | 0,9 | 27,8 |
| 12 | 10 | 1,35 | 44,3 | 138 | ‐0,17 | 23,26 | 2,13 | Pré‐púbere | 2,7 | 454 | N/A | 90 | 8,5 | N/A | N/A |
| 13 | 6 | 1,9 | 25,9 | 113,2 | ‐1,44 | 20,21 | 2,56 | Pré‐púbere | N/A | 218 | 187 | 98 | N/A | ‐1,3 | 33,9 |
| 14 | 16 | 0,8 | 99,3 | 164,5 | ‐1,21 | 36,70 | 3,26 | Púbere | 1,5 | 404 | 273 | 94 | 9 | N/A | N/A |
| 15 | 7 | 1,6 | 36,5 | 130 | 0,73 | 21,60 | 2,74 | Pré‐púbere | 1,6 | 192 | N/A | 85 | 3,9 | N/A | N/A |
| 16 | 10 | 0,8 | 51,9 | 140 | 0,21 | 26,50 | 2,73 | Pré‐púbere | 1,8 | 218 | 233 | 69 | 7,1 | N/A | N/A |
| 17 | 4 | 1 | 22,5 | 102,5 | ‐1,14 | 21,42 | 3,19 | Pré‐púbere | 9,8 | 321 | 237 | 25 | 7,6 | N/A | 31,7 |
| 18 | 12 | 1,1 | 56,7 | 136,5 | ‐1,92 | 30,43 | 3,02 | Pré‐púbere | 2,8 | 371 | N/A | 86 | 10,1 | ‐0,6 | 46,2 |
| 19 | 11 | 1,6 | 34,4 | 130 | ‐2,4 | 20,30 | 1,09 | Pré‐púbere | 0,8 | 159 | 129 | 86 | 7,1 | N/A | N/A |
| 20 | 7 | 0,9 | 28,7 | 118,5 | ‐0,98 | 20,44 | 2,47 | Pré‐púbere | 0,5 | 123 | 107 | 93 | 3,6 | 0,6 | 30,6 |
| 21 | 1 | b | 11,9 | 75 | ‐1,29 | 21,16 | 3,08 | Pré‐púbere | 5,3 | 715 | 196 | 93 | 6,6 | N/A | N/A |
Escores z para altura para idade e IMC calculados no Anthro e AnthroPlus da OMS. A determinação da densidade óssea foi feita em um dispositivo Lunar iDXA (GE Healthcare). Escores z para densidade mineral óssea calculados e composição corporal expressa como percentual de gordura corporal (% de GC). Escores z para densidade mineral óssea não calculados no paciente 17 devido à idade < 5 anos. N/A, não aplicável.
Intervalos de referência: lactato, 0,5–2,2 mmol/L; triglicerídeos, ≤ 100mg/dL em idades < 10 anos, ≤ 130mg/dL em idades entre 10‐19 anos, ≤ 150mg/dL em adultos; colesterol total, < 129mg/dL; glicose, 60‐99mg/dL; ácido úrico, 2,4–7mg/dL.
IMC, índice de massa corporal.
. IMC, índice de massa corporal.")
A composição corporal foi analisada em 10 pacientes (oito com GSDIa, dois com GSDIb) (tabela 2). Para avaliação do tamanho do fígado, 14 pacientes foram submetidos à ultrassonografia abdominal; desses, cinco apresentaram tamanho normal do fígado e um deles apresentou um nódulo hepático visível. Os oito pacientes restantes apresentaram hepatomegalia, e dois apresentaram mais de três nódulos detectáveis.
DiscussãoA caracterização da história natural de doenças raras e da eficácia de tratamentos para essas doenças é sempre prejudicada por amostras pequenas.10 O pequeno número de pacientes é atribuível não apenas à raridade dessas doenças, mas também ao subdiagnóstico, principalmente de casos com manifestações clínicas relativamente leves. Portanto, estudos como este – a primeira caracterização de uma população de pacientes com GSDI no Brasil – são primordiais, considerando seu papel fundamental em possibilitar a realização posterior de uma meta‐análise e atingir conclusões mais completas.
O diagnóstico da GSDI foi tardio nessa amostra, confirmando nossa hipótese inicial. De acordo com a literatura, a idade normal no início dos sintomas em pacientes com GSDI é de três meses.4 Este estudo não avaliou a variável “idade no início dos sintomas”, pois acreditamos que ela está sujeita a uma ampla gama de vieses, principalmente de memória. Estudos mostraram que diagnóstico e início do tratamento precoces estão associados a menores chances de complicações.3 Em nossa amostra, o diagnóstico clínico precoce foi estabelecido em um paciente (paciente 5), que desenvolveu sintomas antes do primeiro mês de vida e tinha um irmão mais velho (paciente 6) com diagnóstico confirmado de GSDIa. O último diagnóstico foi aos 132 meses de idade, no paciente 14, que apresentou hipoglicemia subclínica e foi diagnosticado após uma investigação de três anos desencadeada pela baixa estatura do mesmo, representando, assim, um fenótipo de certa forma atenuado da doença. Apesar de a hipoglicemia ser um dos principais sintomas que despertam suspeita clínica de GSDI, às vezes ela pode passar despercebida devido ao uso de ácido láctico como um substrato para metabolismo cerebral.11 Portanto, apesar de a hipoglicemia sintomática ser relatada com mais frequência, sua ausência não determina diagnóstico de GSDI. Em 2003, Shieh et al. publicaram um relato de caso descrevendo o diagnóstico tardio da GSDI e sugeriram que podem ocorrer formas “mais leves” da doença.11 Ainda em 2003, um artigo recomendou que adolescentes com hiperuricemia e hiperlipidemia inexplicadas fossem avaliados para GSDI, mesmo em caso de ausência de hipoglicemia.12
Com relação aos procedimentos de diagnóstico, a maioria dos pacientes em nossa amostra foi submetida à biópsia de fígado.13 Esse achado é muito surpreendente em virtude do aumento mundial na acessibilidade de testes genéticos. O gene G6PC é pequeno (12,5kb, 5 éxons) e, assim, facilmente sequenciado, principalmente porque ele é afetado por mutações comuns na população brasileira, como p.347X e p.R83C, como relatado por Reis et al. em 2011.14 Em nossa amostra, essas mutações foram encontradas em quatro e três dos 10 pacientes com GSDIa, respectivamente (dados não mostrados). Apesar não inteiramente desprovido de risco, a coleta de sangue para testes genéticos é um procedimento bem menos invasivo e dispendioso que a biópsia de fígado para avaliação histopatológica ou de atividade enzimática. A análise histológica isolada do tecido hepático sem mensuração da atividade enzimática não é suficiente para determinar o tipo de GSD, apesar de poder demonstrar deposição de glicogênio e gordura e ser valiosa no diagnóstico diferenciado de outras doenças hepáticas. Por outro lado, ensaios enzimáticos estão disponíveis apenas em muito poucos centros e estão associados a vários desafios de logística, como transporte do tecido (as amostras devem estar preferencialmente frescas ou congeladas) para o laboratório de referência.
Nossos dados sugerem uma tendência de pacientes com maiores escores z para altura para idade e também com maiores escores z para IMC para idade.15 Apesar de essa tendência ter sido afetada por valores discrepantes, ela sugere que manejo dietético intensivo leva a um melhor crescimento à custa de ganho de peso significativo, conforme relatado anteriormente por Weinstein e Wolfsdorf em 2002.6 O tratamento da obesidade em pacientes com GSDI é certamente um tema que merece maior investigação.
O retardo do crescimento é um achado de grande importância em crianças com GSDI,16 e baixa estatura é comum em adultos com a doença. Em nossa amostra, pacientes com controle metabólico inadequado, de acordo com o ESGSD I,4 apresentaram os piores escores z para altura para idade. A fisiopatologia da baixa estatura na GSDI ainda precisa ser elucidada, porém estudos realizados desde 2008 têm mostrado que controle metabólico adequado pode melhorar o crescimento.17,18 Alterações hormonais, variação no pH sanguíneo (devido à acidose metabólica) e hiperlactatemia podem contribuir para esse déficit de crescimento. De acordo com os critérios do ESGSD I,5 metade de todos os pacientes em nossa amostra apresentaram controle metabólico adequado de sua doença, apesar de alguns apresentaram escores z para IMC > 2 SDs, que pode representar, pelo menos em parte, crescimento quase adequado dessa população (18 de 21 pacientes apresentaram escores z para altura para idade > ‐2 SDs).
A finalidade do manejo dietético da GSDI é imitar a produção de glicose endógena. As estratégias de administração exógena de dextrose para manutenção da normoglicemia foram avaliadas e modificadas nos últimos anos. Alimentações frequentes contendo amido parcialmente cozido, alimentação gástrica noturna contínua por gotejamento (CNGDF) de dextrose via tubo nasogástrico e terapia de amido de milho cru (UCCS) são algumas das estratégias disponíveis. Nenhum dos pacientes nesta casuística estava sendo alimentado à noite de forma contínua; todos estavam em terapia de amido de milho cru (cinco ou seis doses por 24 horas, inclusive durante a noite). Uma meta‐análise recente15 comparou vários estudos de UCCS (uso diurno e noturno) a estudos de CNGDF, e constatou melhora em curto e longo prazos do controle metabólico nos pacientes devido à UCCS.19 Portanto, a CNGDF deve ser restrita a casos específicos, pois a inconveniência de ser alimentado por uma bomba e o risco de hipoglicemia severa em caso de interrupção abrupta da alimentação (p. ex. devido à queda de energia ou mau funcionamento da bomba) não superaram os benefícios do controle metabólico da administração intermitente de UCCS durante a noite. Uma fórmula modificada de amido de milho (Glycosade®, Vitaflo, Nestlé Health Nutrition, Vevey, Suíça), e com aprovação concedida pela agência de Administração de Alimentos e Medicamentos (FDA), em 2012, é uma alternativa que pode possibilitar que os pacientes durmam durante a noite.20
A hepatomegalia vista em pacientes com GSDI pode ser resultado de deposição de glicogênio e doença do fígado gorduroso secundária ao aumento no fluxo de ácidos graxos livres do tecido adiposo para o fígado.21 Ultrassom de rotina do fígado é um tipo de diagnóstico não invasivo que pode ser utilizado para avaliar o sucesso em longo prazo do tratamento. Como nossa análise dos achados ultrassonográficos teve como base uma revisão de prontuários, a variabilidade do operador é uma preocupação. A histologia dos adenomas hepatocelulares na GSDI é semelhante à dos adenomas vistos em outras doenças. Várias hipóteses tentaram explicar o desenvolvimento de alterações adenomatosas, como desequilíbrios na proporção de glucagon/insulina, sobrecarga de glicogênio celular e ativação do proto‐oncogene.21 Os três pacientes com adenomas hepáticos em nossa casuística tinham 16, 17 e 25 anos de idade (pacientes 5,6 e 10, respectivamente). Os pacientes 5 e 6 apresentaram controle metabólico inadequado, com hiperuricemia e hipoglicemia apesar de baixos níveis de triglicerídeos e lactato quase normal. O paciente 10, o mais velho em nossa amostra, também apresentou controle metabólico inadequado.
Adenomas hepatocelulares podem ocorrer em 22%‐75% dos adultos com GSDIa, e o risco de transformação maligna é de aproximadamente 10%.21 Como a maioria dos pacientes tinha menos de 20 anos de idade, espera‐se baixa incidência de adenomas nessa população, independentemente do controle metabólico. Até agora, não existem casos de hepatocarcinoma em nossa casuística.
Dos nove pacientes submetidos a exames de densidade óssea, apenas um (paciente 1) apresentou “baixa massa corpórea para a idade cronológica” de acordo com a Declaração de Posição Oficial de 2008da Sociedade Brasileira de Densitometria Óssea.22 Vários mecanismos têm sido sugeridos para explicar a baixa densidade mineral óssea vista em pacientes com GSDI: acidose persistente, perda de cálcio na urina sem substituição adequada, redução da matriz óssea (hipoglicemia leva à diminuição da glicosilação das proteínas da matriz óssea) e alterações nos níveis do hormônio do crescimento (GH).18 Adicionalmente, vários pacientes com GSDI apresentam crescimento puberal anormal,5 e os esteroides sexuais desempenham um importante papel na formação óssea, principalmente durante a puberdade.22 Alguns tecidos devem ser levados em consideração ao analisar a densidade mineral óssea em crianças e adolescentes, como maturação óssea, sexo e fase da puberdade.23 Em geral, os pacientes avaliados nesta casuística apresentaram boa densidade mineral óssea, apesar de sua GSDI.
Apesar da raridade da GSDI, a mesma não pode ser ignorada pelos pediatras. O agente utilizado para tratamento desse erro inato do metabolismo está prontamente disponível em qualquer mercearia ou supermercado. A administração de amido de milho como nutracêutico (alimento utilizado para fins medicinais) em pacientes com GSDI possui o ganho de peso excessivo como efeito colateral, devido ao aumento no consumo total de carboidrato e também provavelmente à inatividade física relativa. Nenhum estudo avaliou a eficácia de exercícios físicos em pacientes com GSDI, e os mesmos não são contraindicados. Portanto, a atividade física, acompanhada de uma prescrição dietética bem formulada considerando os períodos de pré e pós‐exercício, pode ser uma estratégia válida para a administração do ganho de peso em pacientes cujo controle metabólico não é satisfatório.
A maior conscientização dessa doença entre os pediatras deve ajudar na sua busca por um diagnóstico etiológico em casos de hipoglicemia, hepatomegalia, dislipidemia e baixa estatura, que podem ser tratados inadequadamente. O diagnóstico precoce com base em achados clínicos e laboratoriais é viável, fácil e acessível, mesmo em casos de acesso limitado a cuidados especializados. Contudo, o investimento em centros de diagnóstico molecular especializado deve ser garantido e necessário, pois o uso de métodos moleculares praticamente elimina a necessidade de biópsia hepática. O tratamento precoce pode ser instituído em qualquer serviço de saúde, não exige intervenções complexas e reduz o risco de morte, principalmente por prevenir a hipoglicemia severa. Pacientes adequadamente tratados podem ter vidas intelectual e socialmente gratificantes sem limitações, exceto uma dieta especial.
FinanciamentoFIPE‐HCPA, FAPERGSe CNPq.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Este trabalho recebeu o apoio da FIPE‐HCPA, FAPERGS e CNPq. Os autores gostariam de agradecer à equipe multidisciplinar da clínica de Erros Inatos do Metabolismo do Serviço de Genética Médica do HCPA, aos serviços de Gastroenterologia e Hepatologia do HCPA e da PUC, ao SIEM e à Ana Carolina Monteiro, por ajudá‐los neste artigo e com o diagnóstico e atendimento dos pacientes. Gostaríamos de agradecer também ao Dr. Terry Derks pelas oportunidades de aprendizado inestimáveis e ao Dr. David Weinstein por suas diversas lições, que contribuíram direta ou indiretamente para este estudo e que ainda podem se tornar a força motriz para um grupo de pesquisa multicêntrica.
Como citar este artigo: Santos BL, de Souza CF, Schuler‐Faccini L, Refosco L, Epifanio M, Nalin T, et al. Glycogen storage disease type I: clinical and laboratory profile. J Pediatr (Rio J). 2014;90:572–9.
Estudo conduzido na Universidade Federal do Rio Grande do Sul, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brasil.




