

To identify pathogenic genomic imbalances in patients presenting congenital heart disease (CHD) with extra cardiac anomalies and exclusion of 22q11.2 deletion syndrome (22q11.2 DS).
Methods78 patients negative for the 22q11.2 deletion, previously screened by fluorescence in situ hybridization (FISH) and/or multiplex ligation probe amplification (MLPA) were tested by chromosomal microarray analysis (CMA).
ResultsClinically significant copy number variations (CNVs ≥300kb) were identified in 10% (8/78) of cases. In addition, potentially relevant CNVs were detected in two cases (993kb duplication in 15q21.1 and 706kb duplication in 2p22.3). Genes inside the CNV regions found in this study, such as IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4, SOX7, SEMAD6D, FBN1, and LTPB1 are known to participate in cardiac development and could be candidate genes for CHD.
ConclusionThese data showed that patients presenting CHD with extra cardiac anomalies and exclusion of 22q11.2 DS should be investigated by CMA. The present study emphasizes the possible role of CNVs in CHD.
Identificar desequilíbrios genômicos patogênicos em pacientes apresentando Cardiopatias Congênitas (CC) e anomalias extra cardíacas, e exclusão da Síndrome de Deleção 22q11.2 (SD22q11.2).
MétodosUm total de 78 pacientes negativos para a deleção 22q11.2, previamente testados por hibridação in situ com fluorescência (FISH) e/ou amplificação de múltiplas sondas dependentes de ligação (MLPA), foram avaliados por microarray cromossômico (CMA).
ResultadosForam identificadas variações do número de cópias de DNA (CNVs) clinicamente significativas (≥ 300kb) em 10% (8/78) dos casos, além de CNVs potencialmente relevantes em dois casos (duplicação de 993kb em 15q21.1 e duplicação de 706kb em 2p22.3). Genes envolvidos como IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4, SOX7, SEMAD6D, FBN1 e LTPB1 são conhecidos por atuarem no desenvolvimento cardíaco e podem ser genes candidatos a CC.
ConclusãoEstes dados mostram que pacientes apresentando CC, com anomalias extra cardíacas e exclusão da SD22q11.2, devem ser investigados por CMA. Ainda, este estudo enfatiza a possível função das CNVs nas CC.
Congenital heart disease (CHD) is a common malformation affecting approximately six per 1000 live births. It occurs as an isolated trait or related to multiple congenital anomalies, among which the 22q11.2 deletion syndrome (22q11.2 DS) is the most common.1 CHD is the most critical manifestation and represents the major morbimortality factor in 22q11.2 DS, affecting between 74% and 80% of patients. Among the variety of CHDs reported, conotruncal and/or aortic arch defects are the most prevalent.2 The cause of cardiac phenotypic heterogeneity is not known, but there is no evidence of correlation with sex, race, 22q11.2 deletion size, or parental origin of the deletion.3
It is recommended that all newborns or children presenting CHD and dysmorphism or other congenital anomalies be screened for 22q11.2 deletion.4 In addition, genomic imbalances of other chromosomal regions, including 10p12-p15, 4q21-q35, 8p21-p23, 17p13, and 18q21, can be found in patients with clinical suspicion of 22q11.2 DS and without 22q11.2 deletion.5
Currently, the application of chromosomal microarray analysis (CMA) for clinical diagnosis allows the identification of previously undetectable submicroscopic genomic imbalances, bringing new information about the genesis of congenital defects. The current study investigated a group of individuals presenting CHD with extra cardiac anomalies and exclusion of 22q11.2 DS to identify pathogenic genomic imbalances.
Patients and methodsPatientsThe Research Ethics Committee of the University of Campinas approved this study (No. 487/2009 and 433/2010). All participants or their guardians signed the written informed consent. The evaluation included a standardized protocol as part of a multicenter study of the Brazilian Craniofacial Project, and all individuals were seen by a geneticist.
Initially, a group of 108 patients having CHD with extra cardiac anomalies and clinical suspicion of 22q11.2 deletion were screened by fluorescence in situ hybridization (FISH) and/or multiplex ligation probe amplification (MLPA). Only 78 patients (43 males and 35 females) without 22q11.2 deletion were included in the present study.
Among the CHDs in this cohort, tetralogy of Fallot (ToF) was observed in 40%, ventricular septal defect (VSD) in 22%, and atrial septal defect (ASD) in 8% of cases. In 9% of cases, two of these three defects were observed. The remaining 21% of cases presented other cardiac defects such as truncus arteriosus (TA), bicuspid aortic valve (BAV), and patent ductus arteriosus (PDA), among others.
The main clinical features found in these patients, in addition to CHD, were: facial dysmorphisms – 96% (75/78), neurocognitive and behavioral-developmental abnormalities – 65% (51/78), skeletal abnormalities – 58% (45/78), palatal abnormalities – 43% (34/78), immunological abnormalities – 41% (32/78), growth delay and/or feeding abnormalities – 32% (25/78), gastrointestinal anomalies – 23% (18/78), hearing loss – 10% (8/78), urinary tract anomalies – 10% (8/78), eye abnormalities – 9% (7/78), and neurological abnormalities – 5% (4/78).
MethodsChromosomal microarray analyses were performed for all 78 patients. Patient's parents were not tested. For each sample, 250ng of genomic DNA was labeled and hybridized to the CytoScan HD chip (Affymetrix®, USA), according to the recommendations of the manufacturer. QC metrics were evaluated with the Affymetrix® genotyping console software package. Array data were analyzed using a chromosome analysis suite (ChAS; Affymetrix®). Filters of 25 genetic markers for deletion and 50 for duplications were applied, as recommended by the manufacturer, and a minimum segment length of 300kb for both duplications and deletions was also applied (defined as large copy number variations – CNVs). These CNVs were confirmed using the Nexus Copy Number software (BioDiscovery®, USA). CNVs were further compared with an internal control CNV dataset, including 105 controls from the Brazilian general population, designed by using Cytoscan HD (Affymetrix®). Only genomic imbalances found by all the approaches were selected for further investigation, using the following databases: the Database of Genomic Variants (DGV),6 the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER),7 and the Online Mendelian Inheritance in Man (OMIM).8 CNVs considered relevant were carried forward to confirmation by another microarray platform. This confirmation was performed for 18 patients using the Agilent human genome G3 SurePrint 8x60K microarray (Agilent Technologies®, USA). DNA extracted from patients and reference controls (Promega®, USA) were measured for concentration and purity with a Qubit® spectrophotometer (Life Technologies®, USA). The quantity of 500ng of DNA from the patient and of a sex-matched reference DNA were processed for labeling and hybridization according to the manufacturer's protocol (Agilent oligonucleotide array-based CGH for genomic DNA analysis – Enzymatic labeling protocol v.7.3). Slides were scanned in a dual-laser scanner G4900DA (Agilent Technologies®, USA) and the images were extracted and analyzed by using Agilent feature extraction software (v.11.5.1.1). Array data were analyzed using Agilent cytogenomics software (v.3.0.1.1). A flow chart with the patient population that was studied is summarized in Fig. 1.

Flow chart of the patient population and the techniques applied. FISH, fluorescence in situ hybridization; MLPA, multiplex ligation probe amplification; CMA, chromosomal microarray; CNVs, copy number variations; ChAS, chromosome analysis suite; DGV, database of genomic variants; DECIPHER, database of chromosomal imbalance and phenotype in humans using ensemble resources; OMIM, Online Mendelian Inheritance In Man.
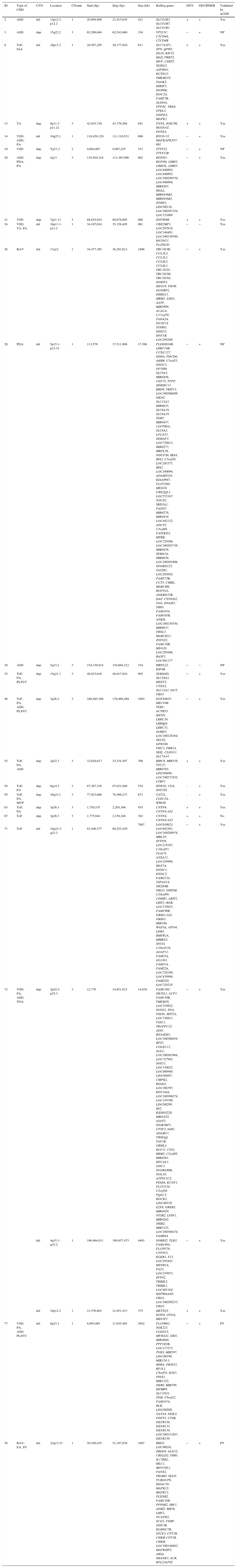
A total of 67 CNVs ≥300kb were detected, comprising 33 deletions and 34 duplications. CNVs with less than 100% of overlap of DECIPHER cases and CNVs with more than four occurrences in the DGV (overlapping more than 50%) were not considered potentially relevant.
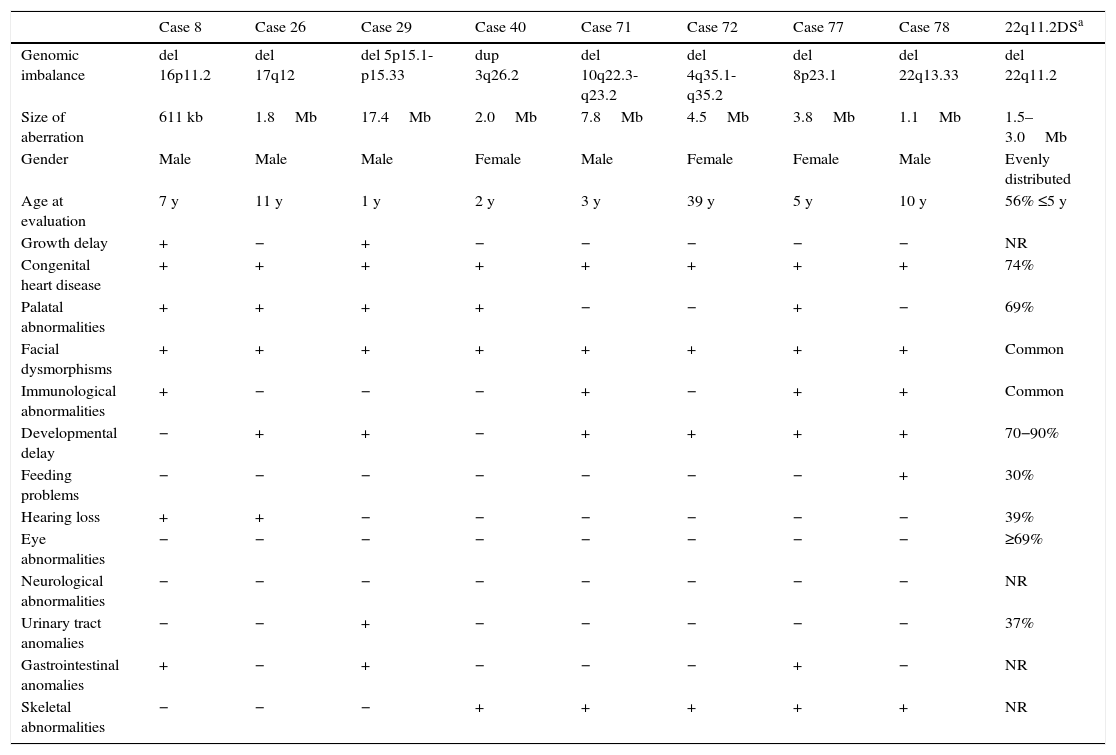
Among 67 CNVs ≥300kb, 25 encompassed genes; 11 deletions and 14 duplications. Eighteen CNVs were confirmed by another microarray platform and two had been previously detected by FISH and/or MLPA techniques (Table 1). Syndromes of multiple congenital anomalies have already been reported in the literature involving eight of these CNVs: 16p11.2 deletion syndrome OMIM 611913 (case 8), 17q12 deletion syndrome OMIM 614527 (case 26), 5p deletion syndrome OMIM 123450 (case 29), 3q duplication syndrome (case 40), 10q22-q23 deletion syndrome OMIM 612242 (case 71), 4q deletion syndrome (case 72), 8p23.1 deletion syndrome (case 77), and 22q13.33 deletion syndrome OMIM 606232 (case 78). These patients had clinical features that overlap with 22q11.2 DS (Table 2).
Type of CHD and CNVs ≥ 300kb observed in the cohort.
| ID | Type of CHD | CNV | Location | CNstate | Start (bp) | Stop (bp) | Size (kb) | RefSeq genes | DGV | DECIPHER | Validated by aCGH |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | ASD | del | 12p12.1-p12.2 | 1 | 20,994,908 | 21,415,634 | 421 | SLCO1B3, SLCO1B7, SLCO1B1 | + | + | Yes |
| 3 | ASD | dup | 15q22.2 | 3 | 62,209,640 | 62,543,660 | 334 | VPS13C, C2CD4A, C2CD4B | − | + | NP |
| 8 | ToF, IAA | del | 16p11.2 | 1 | 29,567,295 | 30,177,916 | 611 | SLC7A5P1, SPN, QPRT, ZG16, KIF22, MAZ, PRRT2, MVP, CDIPT, SEZ6L2, ASPHD1, KCTD13, TMEM219, TAOK2, HIRIP3, INO80E, DOC2A, FAM57B, ALDOA, PPP4C, TBX6, YPEL3, GDPD3, MAPK3 | + | + | Yes |
| 13 | TA | dup | 8p11.1-p11.21 | 3 | 42,935,729 | 43,776,564 | 841 | FNTA, SGK196, HGSNAT, POTEA | + | + | Yes |
| 14 | VSD, ASD, PA | del | 10q25.1 | 1 | 110,420,129 | 111,116,531 | 696 | RNU6-53, MAPKAPK5P1-001 | − | + | Yes |
| 19 | VSD | dup | Yp11.2 | 2 | 9,664,007 | 9,997,425 | 333 | TTTY23, TTTY23B | − | + | NP |
| 20 | ASD, PDA, PA | dup | 2q13 | 3 | 110,504,318 | 111,365,996 | 862 | RGPD5, RGPD6, LIMS3, LIMS3L, LIMS3-LOC440895, LOC440895, LOC100288570, LOC440894, MIR4267, MALL, MIR4436B2, MIR4436B1, NPHP1, LINC00116, LOC100507334, LOC151009 | − | + | Yes |
| 21 | VSD | dup | 7q21.13 | 3 | 88,819,024 | 89,678,695 | 860 | ZNF804B | + | + | Yes |
| 24 | VSD, TA, PA | del | 16p11.1-p11.2 | 1 | 34,197,616 | 35,158,405 | 961 | UBE2MP1, LOC283914, LOC146481, LOC100130700, RN5S411, FLJ26245 | − | − | Yes |
| 26 | BAV | del | 17q12 | 1 | 34,477,385 | 36,283,612 | 1806 | TBC1D3B, CCL3L3, CCL3L1, CCL4L2, CCL4L1, TBC1D3C, TBC1D3H, TBC1D3G, ZNHIT3, MYO19, PIGW, GGNBP2, DHRS11, MRM1, LHX1, AATF, MIR2909, ACACA, C17orf78, TADA2A, DUSP14, SYNRG, DDX52, HNF1B, LOC284100 | − | + | Yes |
| 29 | PDA | del | 5p15.1-p15.33 | 1 | 113,576 | 17,511,896 | 17,398 | PLEKHG4B, LRRC14B, CCDC127, SDHA, PDCD6, AHRR, C5orf55, EXOC3, PP7080, SLC9A3, MIR4456, CEP72, TPPP, ZDHHC11, BRD9, TRIP13, LOC100506688, NKD2, SLC12A7, MIR4635, SLC6A19, SLC6A18, TERT, MIR4457, CLPTM1L, SLC6A3, LPCAT1, SDHAP3, LOC728613, MIR4277, MRPL36, NDUFS6, IRX4, IRX2, C5orf38, LOC285577, IRX1, LOC340094, ADAMTS16, KIAA0947, FLJ33360, MED10, UBE2QL1, LOC255167, NSUN2, SRD5A1, PAPD7, MIR4278, MIR4454, LOC442132, ADCY2, C5orf49, FASTKD3, MTRR, LOC729506, LOC100505738, MIR4458, SEMA5A, MIR4636, LOC100505806, SNORD123, TAS2R1, LOC285692, FAM173B, CCT5, CMBL, MARCH6, ROPN1L, ANKRD33B, DAP, CTNND2, TAG, DNAH5, TRIO, FAM105A, FAM105B, ANKH, LOC100130744, MIR4637, FBXL7, MARCH11, ZNF622, FAM134B, MYO10, LOC285696, BASP1, LOC401177 | − | + | NP |
| 30 | ASD | dup | 5q33.2 | 3 | 154,330,014 | 154,664,212 | 334 | MRPL22, KIF4B | − | − | NP |
| 35 | ToF, PA, PLSVC | dup | 15q21.1 | 3 | 48,023,616 | 49,017,024 | 993 | SEMA6D, SLC24A5, MYEF2, CTXN2, SLC12A1, DUT, FBN1 | − | + | Yes |
| 40 | ToF, PA, ASD, PLSVC | dup | 3q26.2 | 3 | 168,485,398 | 170,468,489 | 1983 | EGFEM1P, MECOM, TERC, ACTRT3, MYNN, LRRC34, LRRIQ4, LRRC31, SAMD7, LOC100128164, SEC62, GPR160, PHC3, PRKCI, SKIL, CLDN11, SLC7A14 | − | + | Yes |
| 52 | ToF, PA, ASD | dup | 2p22.3 | 3 | 32,628,617 | 33,334,307 | 706 | BIRC6, MIR558, TTC27, MIR4765, LINC00486, LOC100271832, LTBP1 | + | + | Yes |
| 58 | ToF, PA | dup | 6q14.3 | 3 | 87,387,316 | 87,921,049 | 534 | HTR1E, CGA, ZNF292 | - | + | Yes |
| 59 | ToF, PA, MVP | dup | 16q23.1 | 3 | 77,923,068 | 78,596,237 | 673 | VAT1L, CLEC3A, WWOX | - | + | Yes |
| 63 | ToF, PA | dup | 3p26.3 | 3 | 1,750,535 | 2,205,306 | 455 | CNTN4, CNTN4-AS2 | + | + | Yes |
| 67 | ToF | dup | 3p26.3 | 3 | 1,775,844 | 2,158,248 | 382 | CNTN4, CNTN4-AS2 | + | + | No |
71 | ToF | del | 10q22.3-q23.2 | 1 | 81,446,577 | 89,253,430 | 7807 | LOC650623, LOC642361, LOC100288974, MBL1P, SFTPD, LOC219347, C10orf57, PLAC9, ANXA11, LOC439990, MAT1A, DYDC1, DYDC2, FAM213A, TSPAN14, SH2D4B, NRG3, GHITM, C10orf99, CDHR1, LRIT2, LRIT1, RGR, LOC170425, FAM190B, GRID1-AS1, GRID1, MIR346, WAPAL, OPN4, LDB3, BMPR1A, MMRN2, SNCG, C10orf116, AGAP11, FAM25A, GLUD1, FAM35A, FAM22A, LOC728190, LOC439994, FAM22D, LOC728218 | − | + | Yes |
| 72 | VSD, PA, ASD, TGA | dup | 2p24.3-p25.3 | 3 | 12,770 | 14,851,012 | 14,838 | FAM110C, SH3YL1, ACP1, FAM150B, TMEM18, LOC339822, SNTG2, TPO, PXDN, MYT1L, LOC730811, TSSC1, TRAPPC12, ADI1, RNASEH1, LOC100506054, RPS7, COLEC11, ALLC, LOC100505964, LOC727982, SOX11, LOC150622, LOC400940, LINC00487, CMPK2, RSAD2, LOC386597, RNF144A, LOC100506274, LOC339788, LINC00299, ID2, KIDINS220, MBOAT2, ASAP2, ITGB1BP1, CPSF3, IAH1, ADAM17, YWHAQ, TAF1B, GRHL1, KLF11, CYS1, RRM2, C2orf48, MIR4261, HPCAL1, ODC1, SNORA80B, NOL10, ATP6V1C2, PDIA6, KCNF1, FLJ33534, C2orf50, PQLC3, ROCK2, LINC00570, E2F6, GREB1, MIR4429, NTSR2, LPIN1, MIR4262, TRIB2, MIR3125, LOC100506474, FAM84A | − | + | Yes |
| del | 4q35.1-q35.2 | 1 | 186,464,011 | 190,957,473 | 4493 | SORBS2, TLR3, FAM149A, FLJ38576, CYP4V2, KLKB1, F11, LOC285441, MTNR1A, FAT1, LOC339975, ZFP42, TRIML2, TRIML1, LOC401164, HSP90AA4P, FRG1, LOC100288255, FRG2 | − | + | Yes | ||
| del | 16p12.2 | 1 | 21,576,802 | 21,951,415 | 375 | METTL9, IGSF6, OTOA, RRN3P1 | + | + | Yes | ||
| 77 | VSD, PA, ASD, PLSVC | del | 8p23.1 | 1 | 8,093,065 | 11,935,465 | 3842 | FLJ10661, SGK223, CLDN23, MFHAS1, ERI1, MIR4660, PPP1R3B, LOC157273, TNKS, MIR597, LINC00599, MIR124-1, MSRA, PRSS55, RP1L1, C8orf74, SOX7, PINX1, MIR1322, XKR6, MIR598, MTMR9, SLC35G5, TDH, C8orf12, FAM167A, BLK, LINC00208, GATA4, NEIL2, FDFT1, CTSB, DEFB136, DEFB135, DEFB134, LOC100133267, DEFB130 | − | + | PV |
| 78 | BAV, EA, SV | del | 22q13.33 | 1 | 50,100,435 | 51,197,838 | 1097 | BRD1, LOC90834, ZBED4, ALG12, CRELD2, PIM3, IL17REL, MLC1, MOV10L1, PANX2, TRABD, SELO, TUBGCP6, HDAC10, MAPK12, MAPK11, PLXNB2, FAM116B, PPP6R2, SBF1, ADM2, MIOX, LMF2, NCAPH2, SCO2, TYMP, ODF3B, KLHDC7B, SYCE3, CPT1B, CHKB-CPT1B, CHKB, LOC100144603, MAPK8IP2, ARSA, SHANK3, ACR, RPL23AP82 | − | + | PV |
ID, patient identification; CHD, congenital heart disease; ASD, atrial septal defect; ToF, tetralogy of Fallot; IAA, interrupted aortic arch; TA, truncus arteriosus; VSD, ventricular septal defect; PA, pulmonary atresia; PDA, patent ductus arteriosus; BAV, bicuspid aortic valve; PLSVC, persistent left superior vena cava; MVP, mitral valve prolapse; TGA, transposition of the great arteries; BAV, bicuspid aortic valve; EA, ectasia of the aorta; SV, sinus of Valsalva; CNV, copy number variation; del, deletion; dup, duplication; bp, base pairs; kb, kilobases; DGV, Database of Genomic Variants;6 DECIPHER, Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources;7 +, presence; −, absence; aCGH, array comparative genomic hybridization (using Agilent Human Genome G3 SurePrint 8x60K Microarray); NP, not performed; PV, previously validated (detected by fluorescence in situ hybridization and/or multiplex ligation probe amplification techniques).
Clinical features of patients presenting CNVs related to syndromes with multiple congenital anomalies that overlap with 22q11.2 DS.
| Case 8 | Case 26 | Case 29 | Case 40 | Case 71 | Case 72 | Case 77 | Case 78 | 22q11.2DSa | |
|---|---|---|---|---|---|---|---|---|---|
| Genomic imbalance | del 16p11.2 | del 17q12 | del 5p15.1-p15.33 | dup 3q26.2 | del 10q22.3-q23.2 | del 4q35.1-q35.2 | del 8p23.1 | del 22q13.33 | del 22q11.2 |
| Size of aberration | 611 kb | 1.8Mb | 17.4Mb | 2.0Mb | 7.8Mb | 4.5Mb | 3.8Mb | 1.1Mb | 1.5–3.0Mb |
| Gender | Male | Male | Male | Female | Male | Female | Female | Male | Evenly distributed |
| Age at evaluation | 7 y | 11 y | 1 y | 2 y | 3 y | 39 y | 5 y | 10 y | 56% ≤5 y |
| Growth delay | + | − | + | − | − | − | − | − | NR |
| Congenital heart disease | + | + | + | + | + | + | + | + | 74% |
| Palatal abnormalities | + | + | + | + | − | − | + | − | 69% |
| Facial dysmorphisms | + | + | + | + | + | + | + | + | Common |
| Immunological abnormalities | + | − | − | − | + | − | + | + | Common |
| Developmental delay | − | + | + | − | + | + | + | + | 70−90% |
| Feeding problems | − | − | − | − | − | − | − | + | 30% |
| Hearing loss | + | + | − | − | − | − | − | − | 39% |
| Eye abnormalities | − | − | − | − | − | − | − | − | ≥69% |
| Neurological abnormalities | − | − | − | − | − | − | − | − | NR |
| Urinary tract anomalies | − | − | + | − | − | − | − | − | 37% |
| Gastrointestinal anomalies | + | − | + | − | − | − | + | − | NR |
| Skeletal abnormalities | − | − | − | + | + | + | + | + | NR |
del, deletion; dup, duplication; kb, kilobases; Mb, megabases; y, years; +, presence; −, absence; NR, not reported; 22q11.2 DS, 22q11.2 deletion syndrome.
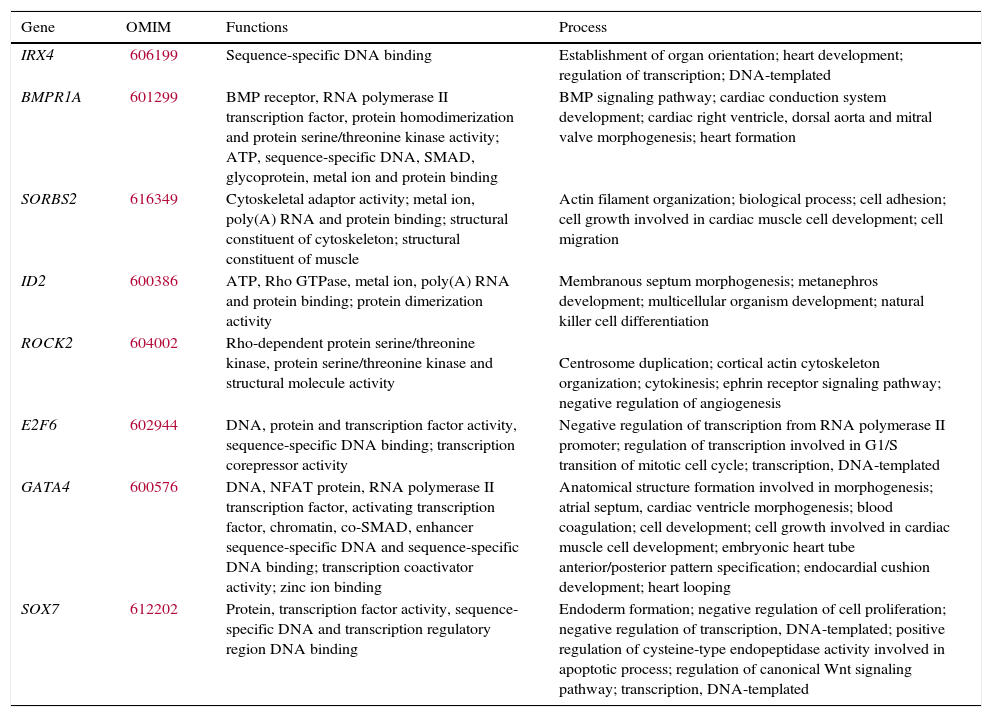
Therefore, pathogenic CNVs were detected in eight out of 78 patients (10%), with sizes from 611kb to 17.4Mb. Genes recognized to participate in cardiac development and candidate genes for CHD are reported in Table 3. In addition, two potentially relevant CNVs encompassing cardiac genes were identified (993kb duplication in 15q21.1 and 706kb duplication in 2p22.3).
Candidate genes for CHD involved in clinically significant CNVs detected.
| Gene | OMIM | Functions | Process |
|---|---|---|---|
| IRX4 | 606199 | Sequence-specific DNA binding | Establishment of organ orientation; heart development; regulation of transcription; DNA-templated |
| BMPR1A | 601299 | BMP receptor, RNA polymerase II transcription factor, protein homodimerization and protein serine/threonine kinase activity; ATP, sequence-specific DNA, SMAD, glycoprotein, metal ion and protein binding | BMP signaling pathway; cardiac conduction system development; cardiac right ventricle, dorsal aorta and mitral valve morphogenesis; heart formation |
| SORBS2 | 616349 | Cytoskeletal adaptor activity; metal ion, poly(A) RNA and protein binding; structural constituent of cytoskeleton; structural constituent of muscle | Actin filament organization; biological process; cell adhesion; cell growth involved in cardiac muscle cell development; cell migration |
| ID2 | 600386 | ATP, Rho GTPase, metal ion, poly(A) RNA and protein binding; protein dimerization activity | Membranous septum morphogenesis; metanephros development; multicellular organism development; natural killer cell differentiation |
| ROCK2 | 604002 | Rho-dependent protein serine/threonine kinase, protein serine/threonine kinase and structural molecule activity | Centrosome duplication; cortical actin cytoskeleton organization; cytokinesis; ephrin receptor signaling pathway; negative regulation of angiogenesis |
| E2F6 | 602944 | DNA, protein and transcription factor activity, sequence-specific DNA binding; transcription corepressor activity | Negative regulation of transcription from RNA polymerase II promoter; regulation of transcription involved in G1/S transition of mitotic cell cycle; transcription, DNA-templated |
| GATA4 | 600576 | DNA, NFAT protein, RNA polymerase II transcription factor, activating transcription factor, chromatin, co-SMAD, enhancer sequence-specific DNA and sequence-specific DNA binding; transcription coactivator activity; zinc ion binding | Anatomical structure formation involved in morphogenesis; atrial septum, cardiac ventricle morphogenesis; blood coagulation; cell development; cell growth involved in cardiac muscle cell development; embryonic heart tube anterior/posterior pattern specification; endocardial cushion development; heart looping |
| SOX7 | 612202 | Protein, transcription factor activity, sequence-specific DNA and transcription regulatory region DNA binding | Endoderm formation; negative regulation of cell proliferation; negative regulation of transcription, DNA-templated; positive regulation of cysteine-type endopeptidase activity involved in apoptotic process; regulation of canonical Wnt signaling pathway; transcription, DNA-templated |
OMIM, Online Mendelian Inheritance In Man.8
Elucidating the genetic contribution of complex disorders is difficult due to the large number, low frequency, and variable effect of predisposition loci. Genomic CNVs have been established as a major source of human genetic variation underlying several multiple congenital anomaly syndromes, including 22q11.2 DS. Determining whether a CNV contributes to a phenotype depends on various factors, including the mode of inheritance, gene content, the copy number state (a loss or a gain), the array platform used, and if it was found in controls from the general population.9 In general, larger CNVs have a higher potential of being pathogenic, as it is more possible that a dosage-sensitive gene is involved, and/or a larger number of genes are included in the imbalance, resulting in an abnormal phenotype.10
The classification of a CNV not reported before, and not including a previously known disease gene, can vary between different populations.10 Therefore, the present CNV analysis was conducted focusing on diagnosis and according to three analytical approaches, as there is not a Brazilian control population database available yet. A minimum size of 300kb was considered (defined as large CNVs), the number of genetic markers recommended, and only CNVs identified by all three approaches were selected for research and confirmation.
Pathogenic CNVs were detected in eight out of 78 patients (10%) and genes involved such as IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4, and SOX7 are known to participate in cardiac development and would be candidate genes for CHD.11–18
Case 8 showed a 611kb deletion overlapping the critical region of 16p11.2 deletion syndrome. Patients with this syndrome usually present developmental delay, intellectual disability, features of autism spectrum disorders, seizures (epilepsy), and obesity,19 not presented in this patient. Less frequently, CHD, minor facial dysmorphisms, and speech delay were observed,19 in concordance with this patient. This patient had ToF and interrupted aortic arch. This deleted region does not allocate candidate genes related to CHD, to date.
Case 26 showed a 1.8Mb deletion overlapping the critical region of 17q12 deletion syndrome. Deletions of this region, including the HNF1β gene, have been found in patients with maturity-onset diabetes of the young type 5 (MODY5), and in patients with cystic renal disease, renal dilations, pancreatic atrophy, and liver abnormalities,20 not present in this patient. Recently, intellectual disability, speech delay, and facial dysmorphisms were associated with this deletion,21 in common with this patient, but CHD has been not mentioned. This patient had BAV. There are no candidates genes related to CHD reported in this deleted region, to date.
Case 29 presented a 17.4Mb deletion in 5p15.1-p15.33, harboring the 5p deletion syndrome (cri du chat syndrome). Infants with this condition often have a high-pitched cry that sounds like that of a kitten, microcephaly, low birth weight, and hypotonia,22 not reported by patient's parents (evaluated with 1 year). Affected individuals also have intellectual disability and developmental delay, distinctive facial features, including hypertelorism, low-set ears, small jaw, and CHD,22 as observed in the present patient. The low resolution of the previous karyotype [46,XY] hindered the diagnosis of this patient, which was improved using aGH. The patient presents PDA. The deleted region allocates a cardiac transcription factor gene, IRX4, in which mutations were related to CHD.11
Case 40 presented a 2Mb duplication in 3q26.2, not harboring the critical region of 3q duplication syndrome (defined as 3q26.31-q27.3). Recently, cases with partial 3q duplications involving the region of this CNV have been reported and associated with this syndrome.23 The phenotype usually involves CHD, brain abnormalities, microcephaly, and atypical facial dysmorphisms in common with this patient. The patient had ToF, ASD, pulmonary atresia, and persistent left superior vena cava. There are no candidate genes related to CHD reported in this duplicated region, to date.
Case 71 showed a 7.8Mb deletion in 10q22.3-q23.2; this region was recently associated with a new syndrome characterized by behavioral and neurodevelopmental impairment, including cognitive delay, autism, hyperactivity, and psychiatric ilness.24 This patient presented only developmental delay. In addition, mild facial dysmorphisms and CHD were observed less frequently in 10q22-q23 deletion syndrome,25 both present in this patient. The patient had ToF. This deleted region harbors the BMPR1A gene, suggested as a candidate gene for CHD because of its role in embryonic development and heart formation.12
Case 72 showed a 4.5Mb deletion in 4q35.1-q35.2. Deletions in 4q are rare and are associated with intellectual disability, craniofacial dysmorphisms, low-set ears, cleft palate, CHD, and fifth finger anomalies.26 This patient presented most of these characteristics, with exception of low-set ears and cleft palate. In addition, this patient showed a 14.8Mb duplication in 2p24.3-p25.3 and a 375kb duplication in 16p12.2, both CNVs without a specific phenotype described to date. The patient had VSD, ASD, pulmonary atresia, and transposition of the great arteries. The 4q35.1-q35.2 deleted region allocates the SORBS2 gene, which is highly expressed in intercalated discs of normal heart tissues.13 The 2p24.3-p25.3 duplicated region harbors the ID2, ROCK2, and E2F6 genes, which have roles in heart development and are candidate genes for CHD.14–16
Case 77 showed a 3.8Mb deletion harboring the critical region of 8p23.1 deletion syndrome; CHD is the hallmark of this syndrome, mainly atrioventricular septal defects, atrial septal defect, and pulmonary stenosis,5 in common with the findings of this patient. In addition, it has been observed that microcephaly, facial dysmorphisms, developmental delay, and behavioral problems are associated with this syndrome.5 The present patient had VSD, ASD, pulmonary atresia, and persistent left superior vena cava. This deleted region harbors GATA4 and SOX7 genes, both transcription factors that have roles in cardiac embryogenesis. They have been suggested as candidate genes for CHD, based on animal models, mutations reported by patients, and CNVs studies.17,18
Case 78 presented a 1.1Mb deletion encompassing the critical region of 22q13.33 deletion syndrome (Phelan-McDermid syndrome), characterized by developmental delay, delayed or absent speech, and autistic features.27 The present patient shares developmental and speech delay. In addition, less frequently, facial dysmorphisms, recurrent infections, CHD, and difficult feeding28 have been observed, in common with this patient. The patient had BAV, aortic ectasia, and sinus of Valsalva. This deleted region does not allocate candidate genes related to CHD, to date.
CNV studies in patients with similar phenotypes of well-known diseases can contribute to a better knowledge of phenotypes, and to the identification of genes and pathways involved in specific CHDs and other defects.29 The CNVs detected in this study that were previously described in clinically recognizable syndromes can help the clinical management and offer appropriate genetic counseling for these patients and their families. These findings also reinforce the importance of CMA for the diagnosis. In addition, two CNVs herein detected are potentially related to CHD.
Case 35 presented a duplication in 15q21.1 (993kb), including seven genes, two of which should be highlighted. SEMA6D plays an essential role in heart morphogenesis30; FBN1 operates in cardiac development and mutations in this gene have been associated with CHD.31 This CNV is described in four individuals in DECIPHER (286150, 250179, 260222, 278520) who have this variant alone; however, none of them had CHD. This CNV was not detected in the DGV. The present patient had ToF, pulmonary atresia, and persistent left superior vena cava.
Case 52 presented a duplication in 2p22.3 (706kb), including seven genes such as LTPB1, which is the major regulator of the TGFB1 gene and acts in the development of heart valve.32 This CNV has been found in two individuals in DECIPHER (249550, 249780), who present only this variant; however, without CHD. This CNV has been described in four studies in the DGV the (variation_8367, variation_53694, variation_5203, variation_5202). The present patient had ToF, pulmonary atresia, and ASD.
Clinically significant CNVs were identified in 10% of cases, reinforcing that CMA is a reliable technology for patients presenting CHD with extra cardiac anomalies and exclusion of 22q11.2 DS. There is an overlap among the clinical manifestations of various syndromes listed here, although comprising distinct frames. The same applies to the phenotype of the present patients. It reinforces the complex mechanisms involved in human embryogenesis, which implies the correct interaction between genes. As several gene interaction mechanisms have not been completely elucidated, this study emphasizes the possible role of CNVs in CHD.
FundingThis study received financial support by the State of São Paulo Research Foundation (FAPESP) (2008/10596-0, 2008/50421-4, 2009/08756-1, and 2011/23794-7) and the National Council for Scientific and Technological Development (CNPq) (149600/2010-0 and 471422/2011-8). VLGSL is supported by (CNPq 304455/2012-1).
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to acknowledge the patients and their parents for cooperation, and colleagues Priscilla Bernardi, Ana Carolina Xavier, Gabriela Ferraz and, Chong Ae Kim, who referred samples. They thank Ana Paula dos Santos and Benilton de Sa Carvalho as well, for assisting with the technique and analysis. They also acknowledge the Microarray Laboratory at the Brazilian Biosciences National Laboratory (LNBio) in the Brazilian Center for Research in Energy and Materials (CNPEM) and the Laboratory of Molecular Genetics of the Faculty of Medical Sciences - Unicamp. They are indebted to Professor Íscia Lopes-Cendes and Fabio Rossi Torres for collaborating with samples in the control group.
Please cite this article as: Molck MC, Simioni M, Vieira TP, Sgardioli IC, Monteiro FP, Souza J, et al. Genomic imbalances in syndromic congenital heart disease. J Pediatr (Rio J). 2017;93:497–507.
Study conducted at Universidade Estadual de Campinas (UNICAMP), Departamento de Genética Médica, Campinas, SP, Brazil.




