

To identify pathogenic genomic imbalances in patients presenting congenital heart disease (CHD) with extra cardiac anomalies and exclusion of 22q11.2 deletion syndrome (22q11.2 DS).
Methods78 patients negative for the 22q11.2 deletion, previously screened by fluorescence in situ hybridization (FISH) and/or multiplex ligation probe amplification (MLPA) were tested by chromosomal microarray analysis (CMA).
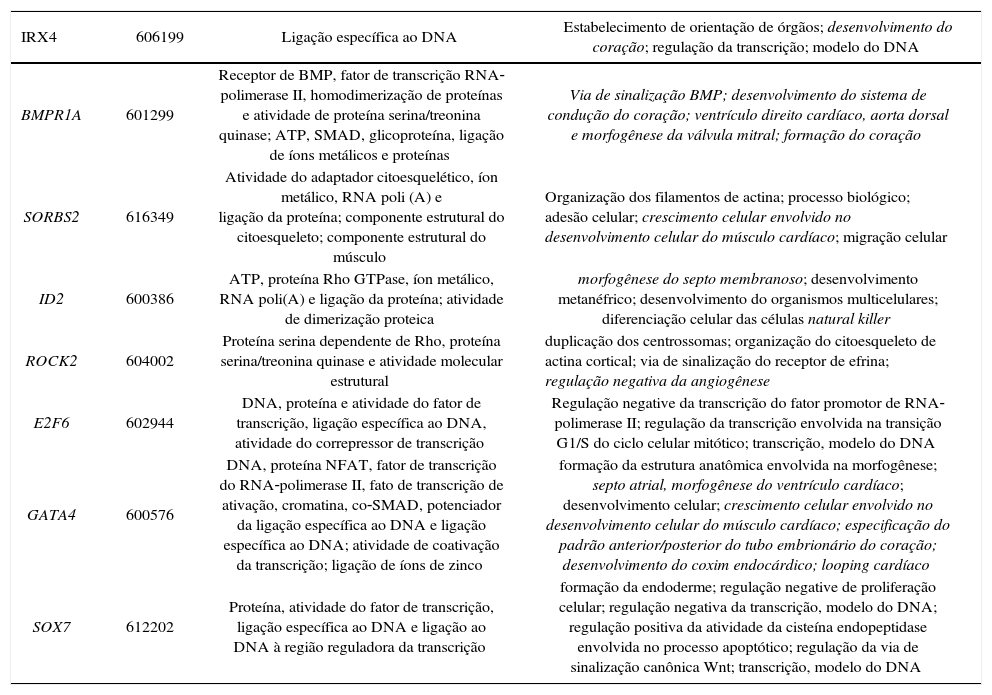
ResultsClinically significant copy number variations (CNVs ≥300kb) were identified in 10% (8/78) of cases. In addition, potentially relevant CNVs were detected in two cases (993kb duplication in 15q21.1 and 706kb duplication in 2p22.3). Genes inside the CNV regions found in this study, such as IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4, SOX7, SEMAD6D, FBN1, and LTPB1 are known to participate in cardiac development and could be candidate genes for CHD.
ConclusionThese data showed that patients presenting CHD with extra cardiac anomalies and exclusion of 22q11.2 DS should be investigated by CMA. The present study emphasizes the possible role of CNVs in CHD.
Identificar desequilíbrios genômicos patogênicos em pacientes que apresentam cardiopatias congênitas (CC) e anomalias extracardíacas e exclusão da síndrome de deleção 22q11.2 (SD22q11.2).
MétodosForam avaliados por microarray cromossômico (CMA) 78 pacientes negativos para a deleção 22q11.2, previamente testados por hibridação in situ com fluorescência (FISH) e/ou amplificação de múltiplas sondas dependentes de ligação (MLPA).
ResultadosForam identificadas variações do número de cópias de DNA (CNVs) clinicamente significativas (≥ 300kb) em 10% (8/78) dos casos, além de CNVs potencialmente relevantes em dois casos (duplicação de 993kb em 15q21.1 e duplicação de 706kb em 2p22.3). Genes envolvidos como IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4, SOX7, SEMAD6D, FBN1 e LTPB1 são conhecidos por atuar no desenvolvimento cardíaco e podem ser genes candidatos a CC.
ConclusãoEsses dados mostram que pacientes que apresentam CC, com anomalias extracardíacas e exclusão da SD22q11.2, devem ser investigados por CMA. Ainda, este estudo enfatiza a possível função das CNVs nas CC.
A cardiopatia congênita (CC) é uma malformação comum que afeta aproximadamente seis de 1.000 nascidos vivos. Ela ocorre isoladamente ou em anomalias congênitas múltiplas, dentre as quais a síndrome de deleção 22q11.2 é a mais comum.1 A CC é a manifestação mais crítica e principal fator de morbimortalidade na síndrome de deleção 22q11.2 (22q11.2DS), afeta entre 74% e 80% dos pacientes. Dentre diversas CCs relatadas, os defeitos conotruncais e/ou do arco aórtico são os mais prevalentes.2 A causa da heterogeneidade fenotípica cardíaca não é conhecida, porém não há provas de correlação com sexo, raça, tamanho da deleção 22q11.2 ou origem parental da deleção.3
Recomenda‐se que todos os recém‐nascidos ou crianças que apresentem CC e dismorfismo ou outras anomalias congênitas sejam examinadas para deleção 22q11.2.4 Além disso, desequilíbrios genômicos de outras regiões cromossômicas, inclusive 10p12‐p15, 4q21‐q35, 8p21‐p23, 17p13 e 18q21, podem ser encontrados em pacientes com suspeita clínica de 22q11.2DS e sem deleção 22q11.2.5
Atualmente, a aplicação da análise cromossômica por microarray (CMA) no diagnóstico clínico possibilita a identificação de desequilíbrios genômicos submicroscópicos indetectáveis, traz novas informações sobre a gênese dos defeitos congênitos. Investigamos um grupo de indivíduos que apresentam CC com anomalias extracardíacas e exclusão da 22q11.2DS para identificar desequilíbrios genômicos patogênicos.
Pacientes e métodosPacientesO Comitê de Ética em Pesquisa da Universidade de Campinas aprovou este estudo (números 487/2009 e 433/2010). Todos os participantes ou seus responsáveis assinaram o consentimento informado por escrito. A avaliação incluiu um protocolo padronizado como parte de um estudo multicêntrico do Projeto Crânio‐Face Brasil e todos os indivíduos foram consultados por um geneticista.
Inicialmente, 108 pacientes que apresentam CC com anomalias extracardíacas e suspeita clínica de 22q11.2 foram examinado por hibridização in situ por fluorescência (FISH) e/ou amplificação de múltiplas sondas dependentes de ligação (MLPA). Apenas 78 pacientes (43 homens e 35 mulheres) sem 22q11.2 foram incluídos no presente estudo.
Dentre as CCs nessa coorte, foi observada tetralogia de Fallot (ToF) em 40%, defeito do septo ventricular (DSV) em 22% e defeito do septo atrial (DAS) em 8% dos casos. Em 9% dos casos foram observados dois desses três defeitos. O restante dos 21% dos casos apresentou outros defeitos cardíacos como truncus arteriosus (TA), válvula aórtica bicúspide (VAB) e persistência do canal arterial (PCA), dentre outros.
As principais características clínicas encontradas nesses pacientes, além de CC, foram: dismorfismos faciais – 96% (75/78), alterações neurocognitivas e comportamentais – 65% (51/78), alterações esqueléticas – 58% (45/78), alterações palatinas – 43% (34/78), alterações imunológicas – 41% (32/78), atraso no crescimento e/ou alterações na alimentação – 32% (25/78), anomalias gastrointestinais – 23% (18/78), perda de audição – 10% (8/78), anomalias no trato urinário – 10% (8/78), alterações visuais – 9% (7/78) e alterações neurológicas – 5% (4/78).
MétodosAs análises cromossômicas por microarray foram feitas em todos os 78 pacientes. Os pais dos pacientes não foram testados. Para cada amostra, 250ng de DNA genômico foram marcados e hibridados no chip CytoScan HD (Affymetrix®, EUA), de acordo com as recomendações do fabricante. Os parâmetros de controle de qualidade foram avaliados com o pacote de software de sistema de genotipagem Affymetrix®. Os dados de array foram analisados com o software de análise cromossômica (ChAS Affymetrix®). Foram aplicados filtros de 25 marcadores genéticos para deleção e 50 para duplicações, conforme recomendado pelo fabricante, e também foi aplicado um comprimento mínimo do segmento de 300kb para duplicações e deleções (definido como amplas variações no número de cópias (CNVs). Essas CNVs foram confirmadas com o software Nexus Copy Number (BioDiscovery®, EUA). As CNVs foram ainda comparadas com um conjunto de dados de CNVs para controle interno, inclusive 105 controles da população geral brasileira designada com o Cytoscan HD (Affymetrix®). Apenas os desequilíbrios genômicos encontrados em todas as abordagens foram selecionados para investigação mais detalhada, com as seguintes bases de dados: Database of Genomic Variants (DGV),6Database of Chromosomal Imbalance and Phenotype in Humans (DECIPHER)7 e Online Mendelian Inheritance in Man (OMIM).8 As CNVs consideradas relevantes foram transferidas para confirmação por outra plataforma de microarray. Essa confirmação foi feita para 18 pacientes, com o microarray de genoma humano G3 SurePrint 8×60Kda Agilent (Agilent Technologies®, EUA). O DNA extraído dos pacientes e os controles de referência (Promega®, EUA) foram medidos para concentração e pureza com o espectrofotômetro Qubit® (Life Technologies®, EUA). A quantidade de 500ng de DNA do paciente e um DNA de referência com correspondência sexual foi processada para rotulagem e hibridização de acordo com o protocolo do fabricante (hibridização genômica comparativa (CGH) – array de oligonucleotídeos da Agilent para análise do DNA genômico – protocolo de rotulagem enzimática v7.3). Os slides foram digitalizados em um scanner dual‐laser G4900DA (Agilent Technologies®, EUA) e as imagens foram extraídas e analisadas com o software de extração de recursos Agilent (v.11.5.1.1). Os dados de array foram analisados com o software de citogenética (v.3.0.1.1). Um fluxograma com a população de pacientes estudada encontra‐se resumido na figura 1.

Fluxograma com a população de pacientes e técnicas aplicadas.FISH, fluorescence in situ hybridization; MLPA, multiplexligation probe amplification; CMA, chromosomal microarray; CNVs, copy number variations; ChAS, chromosome analysis suite; DGV,database of genomic variants; DECIPHER, database of chromosomal imbalance and phenotype in humans using ensemble resources;OMIM, Online Mendelian Inheritance In Man.
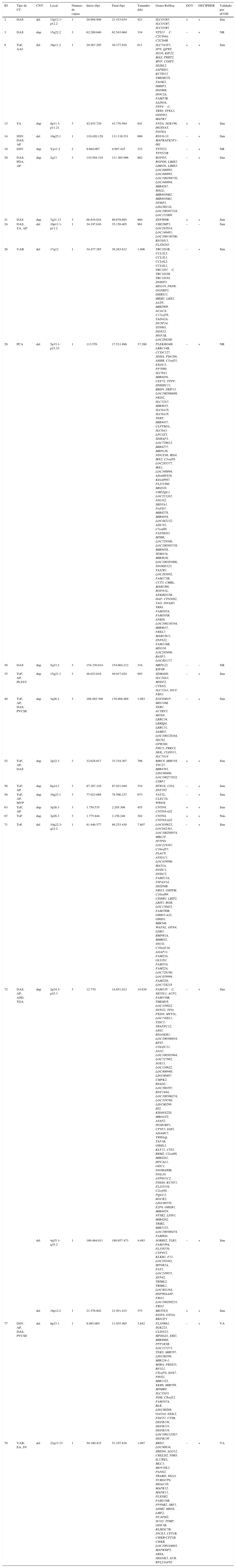
Foram detectadas 67 CNVs ≥ 300kb, 33 deleções e 34 duplicações. As CNVs com menos de 100% de sobreposição de casos na DECIPHER e as CNVs com mais de quatro ocorrências na DGV (sobreposição de mais de 50%) não foram consideradas potencialmente relevantes.
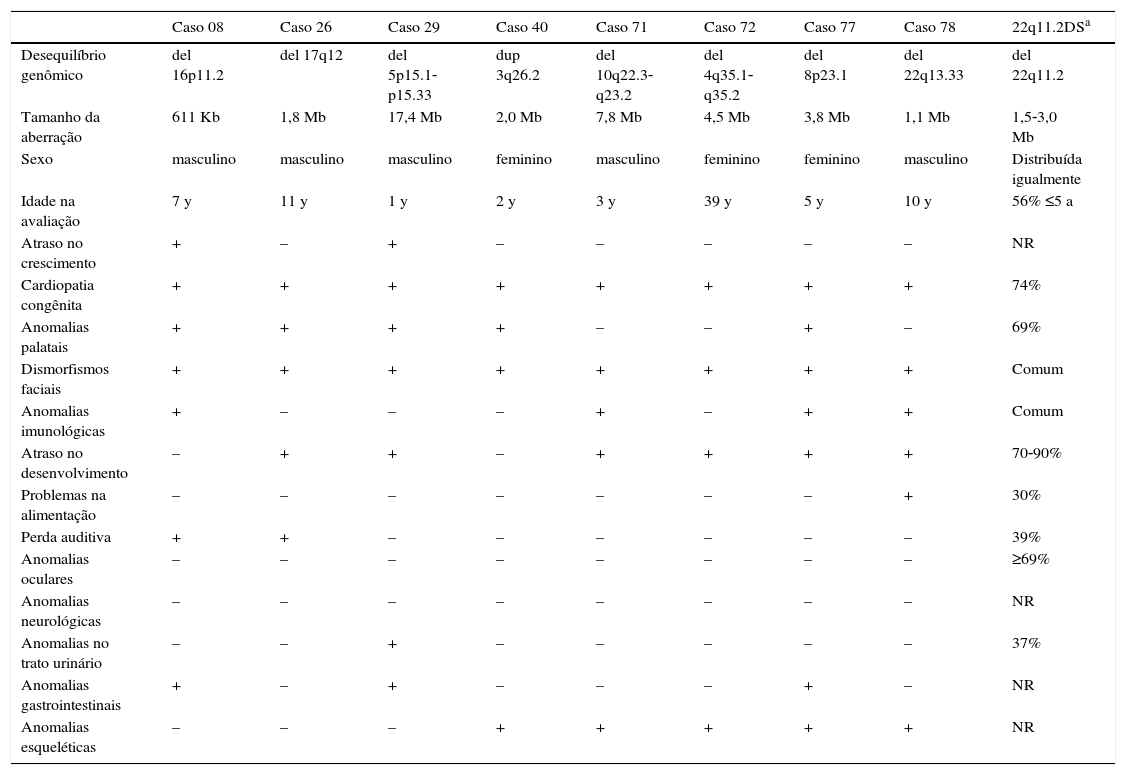
Dentre as 67 CNVs ≥ 300kb, 25 abrangem genes, 11 deleções e 14 duplicações; 18 CNVs foram confirmadas por outra plataforma de microarray e duas foram anteriormente detectadas pelas técnicas de FISH e/ou MLPA (tabela 1). As síndromes de anomalias congênitas múltiplas já foram relatadas na literatura e envolvem oito dessas CNVs: síndrome de deleção 16p11.2 OMIM 611913 (caso 8), síndrome de deleção 17q12 OMIM 614527 (caso 26), síndrome de deleção 5p OMIM 123450 (caso 29), síndrome de duplicação 3q (caso 40), síndrome de deleção 10q22‐q23 OMIM 612242 (caso 71), síndrome de deleção 4q (caso 72), síndrome de deleção 8p23.1 (caso 77) e síndrome de deleção 22q13.33 OMIM 606232 (caso 78). Esses pacientes apresentaram características clínicas que sobrepuseram 22q11.2DS (tabela 2).
Tipo de CC e CNVs ≥ 300kb observado na coorte
| ID | Tipo de CC | CNV | Local | Número de cópias | Início (bp) | Final (bp) | Tamanho (kb) | Genes RefSeq | DGV | DECIPHER | Validado por aCGH |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | DAS | del | 12p12.1‐p12.2 | 1 | 20.994.908 | 21.415.634 | 421 | SLCO1B3, SLCO1B7, SLCO1B1 | + | + | Sim |
| 3 | DAS | dup | 15q22.2 | 3 | 62.209.640 | 62.543.660 | 334 | VPS13C, C2CD4A, C2CD4B | – | + | NR |
| 8 | ToF, AAI | del | 16p11.2 | 1 | 29.567.295 | 30.177.916 | 611 | SLC7A5P1, SPN, QPRT, ZG16, KIF22, MAZ, PRRT2, MVP, CDIPT, SEZ6L2, ASPHD1, KCTD13, TMEM219, TAOK2, HIRIP3, INO80E, DOC2A, FAM57B, ALDOA, PPP4C, TBX6, YPEL3, GDPD3, MAPK3 | + | + | Sim |
| 13 | TA | dup | 8p11.1‐p11.21 | 3 | 42.935.729 | 43.776.564 | 841 | FNTA, SGK196, HGSNAT, POTEA | + | + | Sim |
| 14 | DSV, DAS, AP | del | 10q25.1 | 1 | 110.420.129 | 111.116.531 | 696 | RNU6‐53, MAPKAPK5P1‐001 | – | + | Sim |
| 19 | DSV | dup | Yp11.2 | 2 | 9.664.007 | 9.997.425 | 333 | TTTY23, TTTY23B | – | + | NR |
| 20 | DAS, PDA, AP | dup | 2q13 | 3 | 110.504.318 | 111.365.996 | 862 | RGPD5, RGPD6, LIMS3, LIMS3L, LIMS3‐LOC440895, LOC440895, LOC100288570, LOC440894, MIR4267, MALL, MIR4436B2, MIR4436B1, NPHP1, LINC00116, LOC100507334, LOC151009 | – | + | Sim |
| 21 | DAS | dup | 7q21.13 | 3 | 88.819.024 | 89.678.695 | 860 | ZNF804B | + | + | Sim |
| 24 | DAS, TA, AP | del | 16p11.1‐p11.2 | 1 | 34.197.616 | 35.158.405 | 961 | UBE2MP1, LOC283914, LOC146481, LOC100130700, RN5S411, FLJ26245 | – | – | Sim |
| 26 | VAB | del | 17q12 | 1 | 34.477.385 | 36.283.612 | 1.806 | TBC1D3B, CCL3L3, CCL3L1, CCL4L2, CCL4L1, TBC1D3C, TBC1D3H, TBC1D3G, ZNHIT3, MYO19, PIGW, GGNBP2, DHRS11, MRM1, LHX1, AATF, MIR2909, ACACA, C17orf78, TADA2A, DUSP14, SYNRG, DDX52, HNF1B, LOC284100 | – | + | Sim |
| 29 | PCA | del | 5p15.1‐p15.33 | 1 | 113.576 | 17.511.896 | 17.398 | PLEKHG4B, LRRC14B, CCDC127, SDHA, PDCD6, AHRR, C5orf55, EXOC3, PP7080, SLC9A3, MIR4456, CEP72, TPPP, ZDHHC11, BRD9, TRIP13, LOC100506688, NKD2, SLC12A7, MIR4635, SLC6A19, SLC6A18, TERT, MIR4457, CLPTM1L, SLC6A3, LPCAT1, SDHAP3, LOC728613, MIR4277, MRPL36, NDUFS6, IRX4, IRX2, C5orf38, LOC285577, IRX1, LOC340094, ADAMTS16, KIAA0947, FLJ33360, MED10, UBE2QL1, LOC255167, NSUN2, SRD5A1, PAPD7, MIR4278, MIR4454, LOC442132, ADCY2, C5orf49, FASTKD3, MTRR, LOC729506, LOC100505738, MIR4458, SEMA5A, MIR4636, LOC100505806, SNORD123, TAS2R1, LOC285692, FAM173B, CCT5, CMBL, MARCH6, ROPN1L, ANKRD33B, DAP, CTNND2, TAG, DNAH5, TRIO, FAM105A, FAM105B, ANKH, LOC100130744, MIR4637, FBXL7, MARCH11, ZNF622, FAM134B, MYO10, LOC285696, BASP1, LOC401177 | – | + | NR |
| 30 | DAS | dup | 5q33.2 | 3 | 154.330.014 | 154.664.212 | 334 | MRPL22, KIF4B | – | – | NR |
| 35 | ToF, AP, PLSVC | dup | 15q21.1 | 3 | 48.023.616 | 49.017.024 | 993 | SEMA6D, SLC24A5, MYEF2, CTXN2, SLC12A1, DUT, FBN1 | – | + | Sim |
| 40 | ToF, AP, DAS, PVCSE | dup | 3q26.2 | 3 | 168.485.398 | 170.468.489 | 1.983 | EGFEM1P, MECOM, TERC, ACTRT3, MYNN, LRRC34, LRRIQ4, LRRC31, SAMD7, LOC100128164, SEC62, GPR160, PHC3, PRKCI, SKIL, CLDN11, SLC7A14 | – | + | Sim |
| 52 | ToF, AP, DAS | dup | 2p22.3 | 3 | 32.628.617 | 33.334.307 | 706 | BIRC6, MIR558, TTC27, MIR4765, LINC00486, LOC100271832, LTBP1 | + | + | Sim |
| 58 | ToF, AP | dup | 6q14.3 | 3 | 87.387.316 | 87.921.049 | 534 | HTR1E, CGA, ZNF292 | – | + | Sim |
| 59 | ToF, AP, MVP | dup | 16q23.1 | 3 | 77.923.068 | 78.596.237 | 673 | VAT1L, CLEC3A, WWOX | – | + | Sim |
| 63 | ToF, AP | dup | 3p26.3 | 3 | 1.750.535 | 2.205.306 | 455 | CNTN4, CNTN4‐AS2 | + | + | Sim |
| 67 | ToF | dup | 3p26.3 | 3 | 1.775.844 | 2.158.248 | 382 | CNTN4, CNTN4‐AS2 | + | + | Não |
| 71 | ToF | del | 10q22.3‐q23.2 | 1 | 81.446.577 | 89.253.430 | 7.807 | LOC650623, LOC642361, LOC100288974, MBL1P, SFTPD, LOC219347, C10orf57, PLAC9, ANXA11, LOC439990, MAT1A, DYDC1, DYDC2, FAM213A, TSPAN14, SH2D4B, NRG3, GHITM, C10orf99, CDHR1, LRIT2, LRIT1, RGR, LOC170425, FAM190B, GRID1‐AS1, GRID1, MIR346, WAPAL, OPN4, LDB3, BMPR1A, MMRN2, SNCG, C10orf116, AGAP11, FAM25A, GLUD1, FAM35A, FAM22A, LOC728190, LOC439994, FAM22D, LOC728218 | ‐ | + | Sim |
| 72 | DAS, AP, ASD, TGA | dup | 2p24.3‐p25.3 | 3 | 12.770 | 14.851.012 | 14.838 | FAM110C, SH3YL1, ACP1, FAM150B, TMEM18, LOC339822, SNTG2, TPO, PXDN, MYT1L, LOC730811, TSSC1, TRAPPC12, ADI1, RNASEH1, LOC100506054, RPS7, COLEC11, ALLC, LOC100505964, LOC727982, SOX11, LOC150622, LOC400940, LINC00487, CMPK2, RSAD2, LOC386597, RNF144A, LOC100506274, LOC339788, LINC00299, ID2, KIDINS220, MBOAT2, ASAP2, ITGB1BP1, CPSF3, IAH1, ADAM17, YWHAQ, TAF1B, GRHL1, KLF11, CYS1, RRM2, C2orf48, MIR4261, HPCAL1, ODC1, SNORA80B, NOL10, ATP6V1C2, PDIA6, KCNF1, FLJ33534, C2orf50, PQLC3, ROCK2, LINC00570, E2F6, GREB1, MIR4429, NTSR2, LPIN1, MIR4262, TRIB2, MIR3125, LOC100506474, FAM84A | – | + | Sim |
| del | 4q35.1‐q35.2 | 1 | 186.464.011 | 190.957.473 | 4.493 | SORBS2, TLR3, FAM149A, FLJ38576, CYP4V2, KLKB1, F11, LOC285441, MTNR1A, FAT1, LOC339975, ZFP42, TRIML2, TRIML1, LOC401164, HSP90AA4P, FRG1, LOC100288255, FRG2 | – | + | Sim | ||
| del | 16p12.2 | 1 | 21.576.802 | 21.951.415 | 375 | METTL9, IGSF6, OTOA, RRN3P1 | + | + | Sim | ||
| 77 | DSV, AP, DAS, PVCSE | del | 8p23.1 | 1 | 8.093.065 | 11.935.465 | 3.842 | FLJ10661, SGK223, CLDN23, MFHAS1, ERI1, MIR4660, PPP1R3B, LOC157273, TNKS, MIR597, LINC00599, MIR124‐1, MSRA, PRSS55, RP1L1, C8orf74, SOX7, PINX1, MIR1322, XKR6, MIR598, MTMR9, SLC35G5, TDH, C8orf12, FAM167A, BLK, LINC00208, GATA4, NEIL2, FDFT1, CTSB, DEFB136, DEFB135, DEFB134, LOC100133267, DEFB130 | – | + | VA |
| 78 | VAB, EA, SV | del | 22q13.33 | 1 | 50.100.435 | 51.197.838 | 1.097 | BRD1, LOC90834, ZBED4, ALG12, CRELD2, PIM3, IL17REL, MLC1, MOV10L1, PANX2, TRABD, SELO, TUBGCP6, HDAC10, MAPK12, MAPK11, PLXNB2, FAM116B, PPP6R2, SBF1, ADM2, MIOX, LMF2, NCAPH2, SCO2, TYMP, ODF3B, KLHDC7B, SYCE3, CPT1B, CHKB‐CPT1B, CHKB, LOC100144603, MAPK8IP2, ARSA, SHANK3, ACR, RPL23AP82 | – | + | VA |
‐, ausência; +, presença; AAI, arco aórtico interrompido; aCGH, hibridização genômica comparativa em array (com o microarray de Genoma Humano G3 SurePrint 8×60Kda Agilent); AP, atresia pulmonar; bp, pares de base; CC, cardiopatia congênita; CNV, variação no número de cópias; DAS, defeito do septo atrial; DECIPHER, Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources7; del, exclusão; DGV, Database of Genomic Variants;6 DSV, defeito do septo ventricular; dup, duplicação; EA, ectasia da aorta; ID, identificação do paciente; kb, kilobase; NR, não realizado; PCA, persistência do canal arterial; PVCSE, persistência da veia cava superior esquerda; PVM, prolapso da válvula mitral; SV, seio de valsalva; TA, truncus arteriosus; TGA, transposição das grandes artérias; ToF, tetralogia de Fallot; VA, validado anteriormente (detectado pelas técnicas de FISH e/ou MLPA); VAB, válvula aórtica bicúspide.
Características clínicas dos pacientes que apresentam CNVs relacionadas a síndromes com anomalias congênitas múltiplas que com sobreposição da 22q11.2DS
| Caso 08 | Caso 26 | Caso 29 | Caso 40 | Caso 71 | Caso 72 | Caso 77 | Caso 78 | 22q11.2DSa | |
|---|---|---|---|---|---|---|---|---|---|
| Desequilíbrio genômico | del 16p11.2 | del 17q12 | del 5p15.1‐p15.33 | dup 3q26.2 | del 10q22.3‐q23.2 | del 4q35.1‐q35.2 | del 8p23.1 | del 22q13.33 | del 22q11.2 |
| Tamanho da aberração | 611 Kb | 1,8 Mb | 17,4 Mb | 2,0 Mb | 7,8 Mb | 4,5 Mb | 3,8 Mb | 1,1 Mb | 1,5‐3,0 Mb |
| Sexo | masculino | masculino | masculino | feminino | masculino | feminino | feminino | masculino | Distribuída igualmente |
| Idade na avaliação | 7 y | 11 y | 1 y | 2 y | 3 y | 39 y | 5 y | 10 y | 56% ≤5 a |
| Atraso no crescimento | + | – | + | – | – | – | – | – | NR |
| Cardiopatia congênita | + | + | + | + | + | + | + | + | 74% |
| Anomalias palatais | + | + | + | + | – | – | + | – | 69% |
| Dismorfismos faciais | + | + | + | + | + | + | + | + | Comum |
| Anomalias imunológicas | + | – | – | – | + | – | + | + | Comum |
| Atraso no desenvolvimento | – | + | + | – | + | + | + | + | 70‐90% |
| Problemas na alimentação | – | – | – | – | – | – | – | + | 30% |
| Perda auditiva | + | + | – | – | – | – | – | – | 39% |
| Anomalias oculares | – | – | – | – | – | – | – | – | ≥69% |
| Anomalias neurológicas | – | – | – | – | – | – | – | – | NR |
| Anomalias no trato urinário | – | – | + | – | – | – | – | – | 37% |
| Anomalias gastrointestinais | + | – | + | – | – | – | + | – | NR |
| Anomalias esqueléticas | – | – | – | + | + | + | + | + | NR |
‐, ausência;+,presença; 22q11.2DS, síndrome de deleção 22q11.2; a, anos; del, deleção; dip, duplicação; Kb, kilobase; Mb, megabase;
NR, não relatado,
Portanto, as CNVs patogênicas foram detectadas em oito dos 78 pacientes (10%), com tamanhos de 611 Kb a 17,4 Mb. Os genes que reconhecidamente participam do desenvolvimento cardíaco e que são candidatos a CC são relatados na tabela 3. Além disso, foram identificadas duas CNVs potencialmente relevantes que abrangem genes cardíacos (duplicação de 993kb em 15q21.1 e duplicação de 706kb em 2p22.3).
Genes candidatos a CC envolvidos nas CNVs clinicamente significativas detectadas
| IRX4 | 606199 | Ligação específica ao DNA | Estabelecimento de orientação de órgãos; desenvolvimento do coração; regulação da transcrição; modelo do DNA |
|---|---|---|---|
| BMPR1A | 601299 | Receptor de BMP, fator de transcrição RNA‐polimerase II, homodimerização de proteínas e atividade de proteína serina/treonina quinase; ATP, SMAD, glicoproteína, ligação de íons metálicos e proteínas | Via de sinalização BMP; desenvolvimento do sistema de condução do coração; ventrículo direito cardíaco, aorta dorsal e morfogênese da válvula mitral; formação do coração |
| SORBS2 | 616349 | Atividade do adaptador citoesquelético, íon metálico, RNA poli (A) e ligação da proteína; componente estrutural do citoesqueleto; componente estrutural do músculo | Organização dos filamentos de actina; processo biológico; adesão celular; crescimento celular envolvido no desenvolvimento celular do músculo cardíaco; migração celular |
| ID2 | 600386 | ATP, proteína Rho GTPase, íon metálico, RNA poli(A) e ligação da proteína; atividade de dimerização proteica | morfogênese do septo membranoso; desenvolvimento metanéfrico; desenvolvimento do organismos multicelulares; diferenciação celular das células natural killer |
| ROCK2 | 604002 | Proteína serina dependente de Rho, proteína serina/treonina quinase e atividade molecular estrutural | duplicação dos centrossomas; organização do citoesqueleto de actina cortical; via de sinalização do receptor de efrina; regulação negativa da angiogênese |
| E2F6 | 602944 | DNA, proteína e atividade do fator de transcrição, ligação específica ao DNA, atividade do correpressor de transcrição | Regulação negative da transcrição do fator promotor de RNA‐polimerase II; regulação da transcrição envolvida na transição G1/S do ciclo celular mitótico; transcrição, modelo do DNA |
| GATA4 | 600576 | DNA, proteína NFAT, fator de transcrição do RNA‐polimerase II, fato de transcrição de ativação, cromatina, co‐SMAD, potenciador da ligação específica ao DNA e ligação específica ao DNA; atividade de coativação da transcrição; ligação de íons de zinco | formação da estrutura anatômica envolvida na morfogênese; septo atrial, morfogênese do ventrículo cardíaco; desenvolvimento celular; crescimento celular envolvido no desenvolvimento celular do músculo cardíaco; especificação do padrão anterior/posterior do tubo embrionário do coração; desenvolvimento do coxim endocárdico; looping cardíaco |
| SOX7 | 612202 | Proteína, atividade do fator de transcrição, ligação específica ao DNA e ligação ao DNA à região reguladora da transcrição | formação da endoderme; regulação negative de proliferação celular; regulação negativa da transcrição, modelo do DNA; regulação positiva da atividade da cisteína endopeptidase envolvida no processo apoptótico; regulação da via de sinalização canônica Wnt; transcrição, modelo do DNA |
OMIM, Online Mendelian Inheritance in Man.8
Elucidar a contribuição genética de doenças complexas é difícil devido ao grande número, à baixa frequência e ao efeito variável do local de predisposição. As CNVs genômicas foram estabelecidas como uma importante fonte de variação genética humana inerente a várias síndromes de anomalias congênitas múltiplas, inclusive a 22q11.2DS. Para determinar se a CNV contribui ou não para um fenótipo depende de diversos fatores, inclusive do modo de herança, do conteúdo genético, do número de cópias (perda ou ganho), da plataforma de array usada e se ela foi encontrada em grupos de controle da população geral.9 Em geral, CNVs mais amplas têm um maior potencial de patogenia, pois é mais possível o envolvimento de um gene sensível à dosagem e/ou que um maior número de genes esteja incluído no desequilíbrio, o que resulta em um fenótipo anormal.10
A classificação de uma variação no número de cópias CNV não relatada anteriormente, e que não inclui um gene de doença anteriormente conhecida, pode variar entre diferentes populações.10 Portanto, nossa análise das CNVs foi feita com foco no diagnóstico e de acordo com três abordagens analíticas, pois ainda não há uma base de dados de controle populacional brasileira disponível. Consideramos um tamanho mínimo de 300kb (definido como CNVs amplas), o número recomendado de marcadores genéticos e apenas CNVs identificadas por todas as três abordagens selecionadas para pesquisa e confirmação.
CNVs patogênicas foram detectadas em oito dos 78 pacientes (10%) e os genes envolvidos, como IRX4, BMPR1A, SORBS2, ID2, ROCK2, E2F6, GATA4 e SOX7, são conhecidos por atuar no desenvolvimento cardíaco e seriam candidatos a CC.11–18
O caso 8 mostrou uma deleção de 611kb com sobreposição à região crítica da síndrome de deleção 16p11.2. Os pacientes com essa síndrome normalmente apresentam atraso no desenvolvimento, deficiência intelectual, características de transtornos do espectro autista, convulsões (epilepsia) e obesidade,19 não apresentados por nossos pacientes. Menos frequentemente, foram observados CC, dismorfismos faciais menores e atraso na fala,19 em concordância com nosso paciente. Nosso paciente apresentou ToF e arco aórtico interrompido. Essa região excluída não aloca genes candidatos a CC, até o momento.
O caso 26 mostrou uma deleção de 1,8kb com sobreposição à região crítica da síndrome de deleção 17q12. As deleções dessa região, inclusive o gene HNF1β, foram encontradas em pacientes com diabete tipo MODY 5 (MODY5) e em pacientes com doença renal cística, dilatações renais, atrofia pancreática e anormalidades hepáticas,20 não apresentadas por nosso paciente. Recentemente, deficiência intelectual, atraso na fala e dismorfismos faciais foram associados a deleção,21 compatível com nosso paciente, porém CC não foi mencionada. Nosso paciente apresentou VAB. Não houve relato de genes candidatos a CC até o momento.
O caso 29 apresentou deleção de 17,4 Mb em 5p15.1‐p15.33, abrangeu a síndrome de deleção 5p (síndrome de Cri Du Chat). Os neonatos com essa condição normalmente apresentam grudo agudo que soa como o de um gato, microcefalia, baixo peso ao nascer e hipotonia,22 não relatados pelos pais dos pacientes (avaliado por um ano). Os indivíduos afetados também apresentam deficiência intelectual e atraso no desenvolvimento, características faciais distintivas, inclusive hipertelorismo, orelhas baixas, mandíbula pequena e CC,22 observados em nosso paciente. A baixa resolução do cariótipo anterior [46,XY] prejudicou o diagnóstico desse paciente, que foi aprimorado com hibridação genômica em arrays (aGH). Nosso paciente apresenta PCA. A região excluída aloca um gene com fator de transcrição cardíaco, o IRX4, nas quais as mutações estavam relacionadas a CC.11
O caso 40 apresentou duplicação de 2 Mb em 3q26.2, não abrangeu a região crítica da síndrome de duplicação 3q (definida como 3q26.31‐q27.3). Recentemente, casos com duplicações parciais 3q que envolvem a região de nossa CNV foram relatados e associados a essa síndrome.23 O fenótipo normalmente envolve CC, anomalias cerebrais, microcefalia e dismorfismos faciais atípicos compatíveis com nosso paciente. Nosso paciente apresentou ToF, DAS, atresia pulmonar e persistência da veia cava superior esquerda. Não houve relato de genes candidatos a CC nessa região duplicada até o momento.
O caso 71 mostrou uma deleção de 7,8 Mb em 10q22.3‐q23.2 e essa região foi recentemente associada a uma nova síndrome caracterizada por comprometimento comportamental e do desenvolvimento neurológico, inclusive atraso cognitivo, autismo, hiperatividade e doença psiquiátrica.24 Nosso paciente apresentou apenas atraso no desenvolvimento. Além disso, dismorfismos faciais leves e CC foram observados de forma menos frequente na síndrome de deleção 10q22‐q23,25 ambos apresentados por nosso paciente. Nosso paciente apresentou ToF. Essa região excluída abrange o gene BMPR1A, sugere um gene candidato a CC devido a seu papel no desenvolvimento embrionário e na formação do coração.12
O caso 72 apresentou deleção de 4,5 Mb em 4q35.1‐q35.2. As deleções em 4q são raras e associadas a deficiência intelectual, dismorfismos craniofaciais, orelhas baixas, fissura palatina, CC e anomalias do quinto dedo.26 Nosso paciente apresentou a maior parte dessas características, exceto orelhas baixas e fissura palatina. Além disso, esse paciente mostrou uma duplicação de 14,8 Mb em 2p24.3‐p25.3 e duplicação de 375 bK em 16p12.2, ambas CNVs sem um fenótipo específico descrito até o momento. Nosso paciente apresentou DSV, DAS, atresia pulmonar e transposição das grandes artérias. A região excluída 4q35.1‐q35.2 aloca o gene SORBS2, que é altamente expresso em discos intercalares de tecidos normais do coração.13 A região duplicada de+2p24.3‐p25s abrange os genes ID2, ROCK2 e E2F6, que atuam no desenvolvimento do coração e são genes candidatos a CC.14–16
O caso 77 mostrou uma deleção de 3,8kb que abrangeu a região crítica da síndrome de deleção 8p23.1; a CC é a marca registrada dessa síndrome, principalmente defeitos do septo atrioventricular, defeito do septo atrial e estenose pulmonar,5 compatível com os achados de nosso paciente. Além disso, foi observado que microcefalia, dismorfismos faciais, atraso no desenvolvimento e problemas comportamentais estão associados a essa síndrome.5 Nosso paciente apresentou DSV, DAS, atresia pulmonar e persistência da veia cava superior esquerda. Essa região excluída abrange os genes GATA4 e SOX7, ambos fatores de transcrição que atuam na embriogênese cardíaca. Eles foram sugeridos como os genes candidatos a CC, com base em modelos animais, mutações relatadas por pacientes e estudos sobre CNVs.17,18
O caso 78 apresentou uma deleção de 1,1 Mb que abrangeu a região crítica da síndrome de deleção 22q13.33 (síndrome de Phelan‐McDermid), caracterizada por atraso no desenvolvimento, retardo ou ausência de fala e características autistas.27 Nosso paciente apresenta atraso no desenvolvimento e na fala. Além disso, e de forma menos frequente, foram observados dismorfismos faciais, infecções recorrentes, CC e dificuldade na alimentação,28 compatível com nosso paciente. Nosso paciente apresentou VAB, ectasia da aorta e seio de valsalva. Essa região excluída não aloca genes candidatos a CC até o momento.
Os estudos sobre CNVs em pacientes com fenótipos semelhantes de doenças bem conhecidas podem contribuir para um melhor conhecimento dos fenótipos e para a identificação dos genes e das vias envolvidas em CCs específicas e outras alterações.29 As CNVs detectadas neste estudo anteriormente descritas em síndromes clinicamente reconhecíveis podem ajuda no manejo clínico e proporcionar aconselhamento genético adequado para esses pacientes e suas famílias. Esses achados também reforçam a importância da CMA para o diagnóstico. Além disso, duas CNVs detectadas aqui possivelmente estão relacionadas a CC.
O caso 35 apresentou duplicação em 15q21.1 (993kb), inclusive sete genes, dois dos quais podem ser realçados. O gene SEMA6D atua na morfogênese do coração;30 e o gene FBN1 atua no desenvolvimento do coração e as mutações nesse gene são associadas a CC.31 Essa CNV é descrita em quatro indivíduos na DECIPHER (286150, 250179, 260222, 278520), que apresentam somente essa variação; contudo, nenhum deles apresentou CC. Essa CNV não foi detectada na DGV. Nosso paciente apresentou ToF, atresia pulmonar e persistência da veia cava superior esquerda.
O caso 52 apresentou duplicação em 2p22.3 (706kb), inclusive sete genes como LTPB1, que é o maior regulador do gene TGFB1 e atua no desenvolvimento da válvula do coração.32 Essa CNV foi encontrada em dois indivíduos na DECIPHER (249550, 249780), que apresentaram apenas essa variação; contudo, sem CC. Essa CNV foi descrita em quatro estudos na DGV (Variação_8367, Variação_53694, Variação_5203, Variação_5202). Nosso paciente apresentou ToF, atresia pulmonar e DAS.
CNVs clinicamente significativas foram identificadas em 10% dos casos. Isso reforça que a CMA é uma tecnologia confiável para pacientes que apresentam CC com anomalias extracardíacas e exclusão de 22q11.2DS. Há uma sobreposição entre as manifestações clínicas de várias síndromes listadas aqui, apesar de abranger características distintas. O mesmo aplica‐se ao fenótipo de nossos indivíduos. Isso reforça os mecanismos complexos envolvidos na embriogênese humana, que implica a correta interação entre os genes. Como vários mecanismos de interação entre os genes não foram completamente elucidados, nosso estudo enfatiza a possível atuação das CNVs na CC.
FinanciamentoFundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp) (2008/10596‐0, 2008/50421‐4, 2009/08756‐1 e 2011/23794‐7) e Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (149600/2010‐0 e 471422/2011‐8). VLGSL conta com o apoio de CNPq 304455/2012‐1.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Aos pacientes e seus pais pela cooperação e aos colegas Priscilla Bernardi, Ana Carolina Xavier, Gabriela Ferraz e Chong Ae Kim, que enviaram amostras. A Ana Paula dos Santos e Benilton de Sá Carvalho, pelo apoio na técnica e na análise. Ao Laboratório de Microarray do Laboratório Nacional de Biociências (LNBio) do Centro Nacional de Pesquisa em Energia e Materiais (CNPEM) e ao Laboratório de Genética Molecular da Faculdade de Ciências Médicas – Unicamp. E aos professores Íscia Lopes‐Cendes e Fabio Rossi Torres pela colaboração com amostras em nosso grupo de controle.
Como citar este artigo: Molck MC, Simioni M, Vieira TP, Sgardioli IC, Monteiro FP, Souza J, et al. Genomic imbalances in syndromic congenital heart disease. J Pediatr (Rio J). 2017;93:497–507.
Estudo realizado na Universidade Estadual de Campinas (UNICAMP), Departamento de Genética Médica, Campinas, SP, Brasil.




