



To describe prevalence of associated defects and clinical–genetic characteristics of patients with typical orofacial clefts seen at a reference genetic service.
MethodsDescriptive study conducted between September of 2009 and July of 2014. Two experienced dysmorphologists personally collected and coded clinical data using a validated, standard multicenter protocol. Syndromic cases were defined by the presence of four or more minor defects, one or more major defects, or recognition of a specific syndrome. Fisher's exact and Kruskal–Wallis tests were used for statistics.
ResultsAmong 141 subjects, associated defects were found in 133 (93%), and 84 (59.5%) were assigned as syndromic. Cleft palate was statistically associated with a greater number of minor defects (p<0.0012) and syndromic assignment (p<0.001). Syndromic group was associated with low birth weight (p<0.04) and less access to surgical treatment (p<0.002). There was no statistical difference between syndromic and non-syndromic groups regarding gender (p<0.55), maternal age of 35 years and above (p<0.50), alcohol (p<0.50) and tobacco consumption (p<0.11), consanguinity (p<0.59), recurrence (p<0.08), average number of pregnancies (p<0.32), and offspring (p<0.35).
ConclusionsThere is a lack of information on syndromic clefts. The classification system for phenotype assignment adopted in this study has facilitated recognition of high prevalence of associated defects and syndromic cases. This system may be a useful strategy to gather homogeneous samples, to elect appropriate technologies for etiologic and genotype–phenotype approaches, and to assist with multiprofessional care and genetic counseling.
descrever a prevalência de defeitos associados e as características genético-clínicas de pacientes com fendas orofaciais típicas (FOT) em um serviço de referência em genética.
MétodosEstudo descritivo realizado entre setembro/2009 a julho/2014. Os dados foram colhidos e codificados por dois observadores clínicos com experiência em dismorfologia, utilizando protocolo validado em estudo multicêntrico. Presença de 4 ou mais defeitos minor, um ou mais defeitos major e diagnóstico de síndrome reconhecida foram critérios utilizados para classificar o caso como sindrômico. Utilizou-se Teste Exato de Fisher para análise de variáveis categóricas e Kruskal-Wallis para igualdade de médias.
ResultadosEntre 141 sujeitos, 133 (93%) apresentavam ao menos um defeito minor ou major associado, sendo 84 (59,5%) classificados como sindrômicos. As fendas de palato estiveram associadas com maior número de defeitos minor (p<0,0012) e com a classificação sindrômica (p<0,01). O grupo sindrômico apresentou maior taxa de baixo peso (p<0,04) e menor acesso a tratamento cirúrgico (p<0,02). Não houve diferenças entre os grupos quanto ao gênero (p<0,55), idade materna ≥ 35 anos (p<0,50), ingestão de álcool (p<0,50) e tabagismo (p<0,11), consanguinidade (p<0,59), recorrência familial (p<0,08) e média de gestações (p<0,32) e de filhos nascidos vivos (p<0,35).
ConclusõesExiste escassez de informações sobre fendas sindrômicas. O método de classificação fenotípica utilizado possibilitou a identificação de alta prevalência de defeitos associados e de casos sindrômicos. Este método seria uma alternativa para homogeneizar amostras, determinar tecnologias visando investigação etiológica e estudos de correlação genótipo-fenótipo, além de colaborar para intervenção multiprofissional e aconselhamento genético.
The scientific interest in the typical orofacial clefts (OFCs), represented by paramedian fissures affecting the lip, palate or both, dates back to the mid-eighteenth century.1,2 The development of technological tools in the genomic era has allowed the expansion of knowledge on their etiology, natural history, and risk factors. However, the understanding of overlapping gene–gene and gene–environment associations and their effects on phenotype remains an important challenge. This knowledge forms the theoretical basis upon which cost-effective treatment and prevention proposals should be built.1–4
Typical OFCs are congenital malformations with large epidemiological, social, psychological, and economic impact. The prevalence ranges from one case to every 500–2500 births. This variation reflects the interference of genetic and environmental factors related to ethnic background, geographic region, and nutritional and health status of the population.2–5
In Brazil, there are problems regarding the epidemiological registry of birth defects. In spite of that fact, a recently published estimate predicted the birth of 2900–4000 children with OFCs in the country in 2011.6 Considering the maintenance of the birth rate in the country and taking into account that the treatment of an individual with OFCs extends into adulthood, this estimate of new cases/year has an important economic impact on the health care system.
Surgical repair of the cleft is usually perceived by the family as the only treatment required. However, a patient with a typical OFC requires continued speech therapy, dental, otorhinolaryngological, and psychological support into adulthood, and usually requires more than one surgical intervention.1–4
Longitudinal studies have disclosed other important health-related aspects, highlighting higher mortality rate at any age, higher prevalence of psychiatric disorders, and increased risk for breast, brain, and colon cancer in the individuals and their families. In the United States, a cost of US$ 200,000 per capita has been estimated in the lifelong care of individuals with OFCs, considering specific treatment, control, and prevention of comorbidities.2,3,5
The prevalence of the association of OFCs with other birth defects is a controversial area in the literature. The fact is that, in the presence of associated defects, the OFC is no longer considered an isolated malformation (non-syndromic OFC) with multifactorial etiology, and is then classified as syndromic. It is estimated that it occurs in 30% of cases with cleft lip with or without cleft palate (CL/P) and in 50% of cases of cleft palate (CP).2,3,5,7
Syndromic OFCs may present a known etiology, classified as a monogenic, chromosomal, or teratogenic syndrome, or may present an unknown etiology, constituting cases with multiple malformation, often called multiple congenital defects (MCDs).2,3,5,7,8
The classification of OFCs as syndromic and non-syndromic and the recognition of associated defects and the underlying etiology are important to establish the diagnosis, estimate prognosis, and define therapeutic planning and genetic counseling, all with primary impact on the individual's health. This knowledge is also important to obtain epidemiological information and to increase the power of the genotype–phenotype correlation studies.2,3,5
The objective of this study was to describe the prevalence of associated defects and genetic–clinical characteristics of a cohort of patients with typical OFCs treated at a referral service of clinical genetics.
Materials and methodsThis was a cross-sectional study of subjects with typical OFCs of any age, treated at the Service of Clinical Genetics of Hospital Universitário Professor Alberto Antunes of Universidade Federal de Alagoas (SGC/HUPAA-UFAL), between September of 2009 and July of 2014. Cases of medial, oblique, and submucosal OFCs, as well as bifid uvula were excluded.
Data collection was carried out using the clinical protocol of the Brazilian Database of Clinical and Familial Data.9 The basic complementary assessment included peripheral blood karyotype with GTG banding and resolution of 400 bands for all patients, performed at the Human Cytogenetics Laboratory of Universidade Estadual de Ciências da Saúde de Alagoas.
Malformation screening was performed in specific cases through imaging exams and additional genetic analysis techniques were applied (fluorescence in situ hybridization [FISH], multiplex ligation-dependent probe amplification [MLPA], and array-based genomic hybridization [aGH]), at the Laboratory of Cytogenetics and Human Cytogenomics of the Medical Genetics Department of Universidade Estadual de Campinas.
The classification and codification of OFCs and associated defects were performed by two geneticists experienced in dysmorphology, using the methods described by Monlleó et al.9 The following definitions were used7,8,10–13:
- -
Minor defect: morphological abnormalities that do not imply in significant esthetic or functional impairment.
- -
Syndrome: clinical picture consisting of OFCs associated with minor and/or major defects with previously demonstrated single etiology factor (e.g. chromosomal abnormality) or strongly suspected based on their recurrence in a number of cases.
- -
MCDs: any combination of OFCs with one or more major defects for which no etiological factor has been demonstrated or suspected.
The outcome variable, dependent, was the presence of morphological abnormalities associated with OFCs, with the sample being regrouped according to the phenotypic classification of cases of non-syndromic OFCs (presence of up to three minor defects) and cases of syndromic OFCs (presence of four or more minor defects, diagnosis of the syndrome, and of MCDs). Independent variables and their respective categories were:
Demographic characteristics: age, gender.
Maternal, newborn, and family characteristics: maternal age at conception, maternal level of education, alcohol or tobacco consumption during pregnancy, number of pregnancies and live births of the mother, birth weight (<2500g and ≥2500g), consanguinity, and family history of OFCs.
Clinical characteristics: type of OFCs, severity of cleft lip, type of associated defect, anatomical distribution of major defects and identified syndromes, access to primary surgery for OFCs.
Data were tabulated and analyzed using the programs Microsoft Excel (Microsoft, 2003, Computer Software, WA, USA) and Epi Info™ version 3.5.2. (Epi Info™, GA, USA). Descriptive analysis was performed with frequency distribution, central tendency, and dispersion measures. Fisher's exact test was used for the analysis of categorical variables and the Kruskal–Wallis test for equality of means. The significance level was set at 5% (p<0.05).
The research had the following ethical approvals: 00907/2009-66 (CEP/UFAL), 0009838/2009-56 (CEP/UNCISAL), 059/2008 (CEP/UNICAMP), 14733 (CONEP), and CAAE 35316314.9.1001.5404.
ResultsBetween September of 2009 and July of 2014, a total of 146 patients with OFCs were treated. Of this total, after applying the inclusion criteria, the sample comprised 141 cases.
Age ranged from 0 to 37 years (mean 5±8.48), with 94 (67%) individuals from 0 to 10 years, 34 (24%) between 11 and 20 years, and 13 (9%) older than 20 years of age. Seventy-two (51%) patients were males. Maternal age at conception ranged from 15 to 47 years (mean 24±6.92). Eighteen (15.7%) mothers were adolescents at the time of the pregnancy and 13 (11.3%) were older than 35 years. In 81 (58.7%) cases, the mother had not completed elementary school.
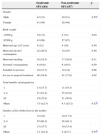
Table 1 shows the behavior of clinical variables regarding the type of OFC. There was a prevalence of CL/P, present in 113 (80%) individuals. These were more common in males, whereas CP was more common in females (p<0.001). Regarding the severity, unilateral clefts occurred in 79 (70%) cases, 53 of which were on the left side, whereas bilateral clefts occurred in 34 (30%) individuals.
Distribution of clinical characteristics of the subjects according to the type of typical orofacial cleft.
| CL/CL/P (113) | CP (28) | p | |
|---|---|---|---|
| Gender | |||
| Male | 65 (57.5) | 7 (25) | 0.0018a |
| Female | 48 (42.5) | 21 (75) | |
| Associated defects | |||
| No defects | 8 (7) | – | |
| Only minor defects | 82 (73) | 18 (64) | 0.10a |
| Minor+major defects | 23 (20) | 10 (36) | |
| Only major defects | – | – | |
| Minor defect | |||
| 1–3 | 48 (46) | 7 (25) | 0.037a |
| ≥4 | 57 (54) | 21 (75) | |
| Mean | 4.2 (±2.8) | 6.5 (±3.3) | 0.0012b |
| Major defect | |||
| 1–3 | 21 (91) | 9 (90) | 0.51a |
| ≥4 | 2 (9) | 1 (10) | |
| Mean | 1.6 (±0.9) | 1.8 (±0.9) | 0.44b |
| Phenotypic classification | |||
| Non-syndromic typical OFCs | 51 (45) | 6 (21) | 0.01a |
| Syndromic OFCs | 62 (55) | 22 (79) | |
CL/CL/P, cleft lip or cleft lip and palate; CP, cleft palate; OFCs, orofacial clefts.
Associated defects of any kind (minor or major) were observed in 105 (93%) cases of CL/P and 28 (100%) of CP. There were no statistically significant differences between the CL/P and CP groups in relation to the type of associated defect (p<0.10); however, the presence of four or more minor defects was higher in the CP group (p<0.0012). The anatomical location, type, and number of individuals with specific major defects are shown in Table 2. The craniofacial region, the cardiovascular system, and the musculoskeletal system were the most frequently affected sites.
Anatomical location, type, and number of subjects with major defects.
| Anatomical site | Type of defects |
|---|---|
| Skull and face | Microcephaly (12), anophthalmia/bilateral microphthalmia, retinal coloboma, bifid nose apex, rudimentary shaped ears (2), iris and corneal hypoplasia, agenesis of teeth, lachrymal duct agenesis, hemifacial microsomia |
| Cardiovascular system | Atrial septal defect (7), ventricular septal defect |
| Musculoskeletal system | Dwarfism with short limbs, arthrogryposis (3), partial absence of lower limb, fused ribs, vertebral segmentation defect |
| Genitourinary tract | Hypospadias, genital ambiguity, renal agenesis, micropenis (2), bilateral cryptorchidism |
| Hands and feet | Syndactyly in hands, hypoplastic distal phalanges of the hands and feet, post-axial polydactyly, terminal transverse defects |
| Skin and annexes | Pterygium (3) |
| Gastrointestinal tract | Esophageal atresia, imperforate anus |
| Central nervous system | Hydrocephaly |
Regarding the phenotypic classification, 84 (59.6%) patients were classified as syndromic OCP, which was more prevalent in the CP group (p<0.01) (Table 1). There were no statistically significant differences between the syndromic and non-syndromic OFC groups regarding the distribution by gender (p<0.55), maternal age risk for chromosomal abnormalities (p<0.50), maternal alcohol intake (p<0.50), maternal smoking (p<0.11), parental consanguinity (p<0.59), familial recurrence (p<0.08), mean number of pregnancies (p<0.32), and mean number of live births (p<0.35). However, the syndromic OFC group showed the highest rate of low birth weight (p<0.04) and less access to surgical treatment (p<0.02) (Table 3).
Distribution of clinical characteristics of the subjects according to the type of typical orofacial cleft.
| Syndromic OFCs(84) | Non-syndromic OFCs(57) | p | |
|---|---|---|---|
| Gender | |||
| Male | 43 (51) | 29 (51) | 0.55a |
| Female | 41 (49) | 28 (49) | |
| Birth weight | |||
| <2500g | 19 (31) | 4 (13) | 0.04 |
| ≥2500g | 43 (69) | 27 (87) | |
| Maternal age ≥35 years | 9 (12) | 4 (10) | 0.50 |
| Maternal alcohol consumption | 22 (26.5) | 14 (25) | 0.50 |
| Maternal smoking | 16 (19.5) | 17 (29.8) | 0.11 |
| Parental consanguinity | 9 (10.8) | 6 (10.5) | 0.59 |
| Familial recurrence | 18 (21.7) | 19 (34) | 0.08 |
| Access to surgical treatment | 46 (54.8) | 41 (71.9) | 0.02 |
| Total number of pregnancies | |||
| 1 | 13 (15.7) | 11 (19.3) | |
| 2–4 | 51 (61.4) | 25 (43.9) | |
| ≥5 | 19 (22.9) | 21 (36.8) | |
| Mean | 3.5 (±2.5) | 4.3 (±3.2) | 0.32b |
| Number of live births born to the mother | |||
| 1 | 15 (18) | 10 (17.5) | |
| 2–4 | 55 (66.3) | 28 (49.1) | |
| ≥5 | 13 (15.7) | 19 (33.4) | |
| Mean | 3.1 (±2.2) | 4 (±3.1) | 0.35b |
OFCs, orofacial clefts.
Of the 84 patients classified as syndromic OFC, 44 (52%) had four or more minor defects, 18 (22%) had one or more major defects constituting cases of MCDs, and 22 (26%) had known syndromes.
Conventional peripheral blood karyotype was performed in 115 individuals (81.5%), which identified abnormalities in seven cases. Of these, four belonged to the syndromic OFC group and had an unbalanced chromosomal abnormality, whereas three had chromosomal polymorphism (pericentric inversion of chromosome 9). Two of the latter met the criteria for the non-syndromic OFC group and one had four minor defects (elbow hyperextensibility, scoliosis, cubitus valgus, genu valgum, and recurvatum), with no nosological diagnosis established.
Seven cases of syndromic OFCs (MCDs) were included in a specific investigation protocol with aGH. One patient showed duplication of 9.2Mb in the region 16p13.2-16p13.3 (46,XX.ish ins(22;16)(q13;p13.2p13.3)), related to the phenotype. Two patients did not have a copy number variation (CNV) and four showed deletions or duplications in regions without known causative genes. Table 4 shows the list of syndromes and laboratory resources used as evidence for diagnosis.
Identified syndromes and respective diagnostic evidence.
| Identified syndrome | n | Type of OFCs | Diagnostic evidence |
|---|---|---|---|
| Chromosome 15 ring syndrome 46,XY,r(15)20 | 1 | CL/P | G band KTP |
| Greig syndrome | 1 | CP | Clinical |
| Del22q11 syndrome: [46,XY.ish del(22)(q11.2q11.2)(TUPLEI-)], | 1 | CL/P | FISH and MLPA |
| Oculofaciocardiodental syndrome | 1 | CL/P | Clinical |
| CHARGE syndrome | 1 | CL/P | Clinical |
| VATER association | 1 | CL/P | Clinical |
| Spondyloepiphyseal dysplasia | 1 | CP | Radiological |
| Waardenburg syndrome | 1 | CL/P | Clinical |
| Escobar syndrome | 1 | CP | Clinical |
| Acrofrontofacionasal dysostosis | 1 | CL/P | Clinical |
| Emanuel syndrome: 47,XX,+der(22)t(11;22)(q23;q11)mat20 | 1 | CP | G band KTP |
| 16p13.3 microduplication syndrome: 46,XX.ish ins(22;16)(q13;p13.2p13.3) | 1 | CP | FISH and aGH |
| Saethre–Chotzen syndrome | 2 | CL/P(2) | Clinical |
| Roberts syndrome | 1 | CL/P | Clinical |
| Goldenhar syndrome | 2 | CP and CL/P | Clinical |
| Ectodermal dysplasia ectrodactyly | 1 | CL/P | Clinical |
| Amniotic band syndrome | 1 | CL/P | Clinical |
| Moebius syndrome | 1 | CP | Clinical |
| Kallmann syndrome | 1 | CL/P | Clinical |
| Mosaic Edwards syndrome: (46,XX/47,XX+18) | 1 | CL/P | G band KTP |
KTP, karyotype; FISH, fluorescence in situ hybridization; MLPA, multiplex ligation-dependent probe amplification; aCGH, array-comparative genomic hybridization; CHARGE syndrome, Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness; VATER association, vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, and radial or renal dysplasia; OFCs, orofacial clefts.
It is a fact that surgical treatment is a crucial stage for the onset of rehabilitation of individuals with OFCs. In this context, it is possible that the genetic evaluation is not seen as necessary at first. However, the identification of associated defects and the etiology has an impact on decision-making regarding both the treatment plan and family genetic counseling.
The World Health Organization (WHO) recommends multidisciplinary care, which includes genetic evaluation. This is based on the performance of a detailed physical examination that takes into account the identification and analysis of dysmorphic signs, allowing the differentiation between syndromic and non-syndromic OFCs.14
Despite the high prevalence, the supply of health care services in the OFC area is still insufficient in many parts of the world. In the period of 1993–2003, there was a significant advance in this area in the Brazilian Unified Health System (SUS). Currently, Brazil has a network of multidisciplinary services accredited by the Ministry of Health for medical-surgical rehabilitation; however, few of them include medical geneticists. Their access is regulated through the National Center of High Complexity Regulation.15
The state of Alagoas has no multidisciplinary services for individuals with OFCs. The primary attention and medium-complexity care in this area also lack infrastructure. As a consequence, many patients have access only to surgical treatment offered by non-governmental organizations (NGOs), which although may bring individual benefits, do not constitute a health policy capable of modifying the scenario of inequity and fragmentation of care.16
As for the diagnostic evaluation and genetic counseling, the SGC/HUPAA-UFAL is the only institution available to meet the SUS demand in the state. The service has maintained a specific outpatient clinic for patients and families with OFCs since 2009.16 The genetic–clinical profile described in this study refers to the patients treated at this clinic.
Demographic and maternal characteristics show a slight predominance of males, broad age range of patients at the time of the initial treatment and mothers at conception, and low maternal level of schooling.
Only one-fourth of the individuals were younger than 1 year of age at the first consultation. Considering the dysmorphic facial aspect of typical OFC cases, it would be expected that most would have been referred for early assessment. The little knowledge of the genetic nature of the OFCs and limited access to genetic services generally observed in Brazil17 may be factors involved.
Almost one-third of the mothers were in the age group of risk (≤18 or ≥35 years) at the time of conception. Teenage pregnancy involves risks related to problems in labor, prematurity, low birth weight, and hypoxia, mainly due to reproductive system immaturity, poor maternal nutrition, and frequent use of legal or illegal drugs.18 Conversely, anomalies caused by numerical chromosomal aberrations significantly increase with advancing age at the time of pregnancy after 35 years.19,20 Low maternal level of schooling, found in most cases of this sample, is another risk factor associated with increased incidence of prematurity, low birth weight, and birth defects in the offspring.18
The predominance of CL/P over CP, of unilateral clefts over bilateral ones, the left side involvement over the right, and the statistical association between CL/P and male gender and CP and female gender corroborate the classic characteristics of OFCs.2–4 Small variations in frequency between subgroups can be justified by the sample size.
The association of OFCs with other malformations has been the subject of several studies worldwide.21–27 Differences in the examiner's experience, patient age, clinical expression variability, availability of technological resources (especially imaging exams), inclusion/exclusion criteria, and type of classification used, among others, are responsible for the observed divergent rates, ranging from 3% to 63%.2–4,7,28
The inclusion of minor defects, the size, and the sample source (Service of Genetics), as well as the clinical assessment by physicians experienced in dysmorphology, can explain the high frequency of associated defects observed in this sample.
Several authors recommend the exclusion of minor defects, considering them an important source of heterogeneity for epidemiological studies. In addition to the acknowledgment that these defects do not imply significant impairment, other reasons for excluding them are the low concordance rates represented by records performed by different professionals and the variability related to sex, ethnicity, and age of onset.10–13
Nonetheless, the predictive value of minor defects has been established, both for the screening of major abnormalities and the diagnostic definition of syndromes more easily recognized by analyzing the set of dysmorphic features, instead of the placing excessive importance on an isolated clinical sign.10–13
Despite the methodological differences between the studies, it is assumed that, in 20% of infants with three or more minor signs, a major sign is detected.10,12 Considering the broad age range and the presence of OFCs as a major sign present in all cases, for this analysis it was decided to establish a cutoff of four or more minor signs to classify the case as potentially syndromic. It is also of note that, in the present study, the clinical observers were the same in all cases (ILM and MIBF), reinforcing the homogeneity of the morphological evaluation.
A high frequency of major and minor defects was observed in the sample, with no preferential association with the OFC type. However, the CP group showed a higher number of associated defects. This result corroborates those found in the literature, as CP has been most often associated with syndromic conditions.2–4
It is noteworthy that all patients with major defects also had minor defects. In these, the most affected anatomical sites were similar to those described in the literature, whose frequency is variable, once again due to the use of different sample selection methods.21–24,27
Considering the phenotypic classification, it was observed that the syndromic OFC group predominated over the non-syndromic group and was statistically associated with CP. The frequency of syndromic OFC cases was higher than that reported in the literature both in the CL/P and CP groups.2–4 These results must be related to the expansion of syndromic case definition criteria used in this study.
The absence of differences between the syndromic and non-syndromic OFC groups regarding the maternal and familial variables probably reflects the etiological heterogeneity of the syndromic group and the sample size. The high frequency of alcohol and tobacco use during pregnancy is alarming and requires health education interventions. Consanguinity and familial recurrence, also important in this sample, are risk factors that should receive the attention of health professionals for an appropriate preventive approach.29,30 The association between syndromic OFCs and low birth weight is expected, considering the higher clinical severity observed in these cases.1,2,4,5
The absence of differences between the syndromic and non-syndromic OFC groups regarding the number of pregnancies may reflect the lack of information by the population about the hereditary nature of OFCs and the difficulty in accessing a genetics service for diagnosis and genetic counseling. The lack of diagnosis, anticipatory conduct, and therapeutic planning provided by the genetic evaluation, as well as the more severe clinical course, may be part of the reasons that justify the significantly lower access to surgical treatment in the syndromic OFC group.
Chromosomal aberrations are traditionally associated with OFCs.2,3,5,7 In this sample, the karyotype clarified the etiology in three cases (46,XY,r(15), 47,XX,+der(22)t(11;22)(q23;q11)mat, and 46,XX/47,+18), and was indicative of the etiology in one [46,XX,add(22)(q13)]. In the latter, the 16p13.3 microduplication syndrome was identified, whose clinical and genetic details will be the object of a specific publication.
In a patient with normal karyotype, the MLPA technique identified the 22q11.2[46,XY.ish del(22)(q11.2q11.2)] deletion, consistent with the clinical suspicion, a result confirmed by FISH. The cases of MCDs with normal karyotype, but that remained without an appropriate diagnosis, reinforce the importance of follow-up and the use of laboratory resources of greater technological sophistication in selected situations. It is noteworthy that among the cases defined as syndromic, most were diagnosed based on clinical criteria, reinforcing the importance of genetic evaluation.
By considering associated defects (minor and major), this study provides a new approach and easy practical application for the codification and classification of OFCs. The proposal extends the concept and establishes the limits between syndromic and non-syndromic OFCs with clinical bases.
In addition to the possibility of planning the clinical-surgical rehabilitation in line with the specific health needs of each individual, the use of this proposal could contribute to homogenize the sample selection and establish the use of new technologies for etiological and genotype–phenotype correlation studies in OFCs. It is expected that this approach will be replicated in studies with larger samples in order to gather information to assess its limitations and potential.
FundingFundação de Amparo à Pesquisa do Estado de Alagoas (FAPEAL), Conselho Nacional de Desenvolvimento Científico e Tecnológico – Programa Interinstitucional de Bolsas de Iniciação Científica (CNPq/PIBIC), and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Monlleó IL, de Barros AG, Fontes MI, de Andrade AK, Brito GM, do Nascimento DL, et al. Diagnostic implications of associated defects in patients with typical orofacial clefts. J Pediatr (Rio J). 2015;91:485–92.
Study conducted at the School of Medicine, Clinical Genetics Service, Hospital Universitário Prof. Alberto Antunes, Universidade Federal de Alagoas (UFAL), Maceió, AL, Brazil.





