


To evaluate the cognitive abilities of children and adolescents with sickle cell anemia diagnosed through neonatal screening and to compare them with healthy controls, adjusting the results to their socioeconomic status.
MethodsCognitive assessment was performed with the Wechsler WISC-III scale in 64 children and adolescents with sickle cell anemia and in 64 controls matched by gender and age, without the disease and without neurological impairment; socioeconomic status was measured by the Criterion Brasil.
ResultsAll cognitive scores were lower in the group of patients. The mean overall IQ, Verbal IQ, and Performance IQ were, respectively, 90.95 for the group of patients and 113.97 for the controls (p<0.001); 91.41 for the group of patients and 112.31 for the controls (p<0.001); 92.34 for the group of patients and 113.38 for the controls (p<0.001). Scores for processing speed, distraction resistance, and perceptual organization were also significantly lower in patients. A direct and significant correlation was detected between socioeconomic status and cognitive scores. In the multivariate analysis, for the same socioeconomic status, a child with sickle cell anemia had an average IQ of 21.2 points lower than the mean IQ observed for the controls (p<0.001), indicating that the disease, adjusted for the socioeconomic effect, is a strong predictor of the overall IQ.
ConclusionThe cognitive impairment of children with sickle cell anemia is severe and manifests even when the disease effect is adjusted to the socioeconomic status. In the authors’ view, such impairment requires an early preventive approach in order to avoid this cognitive damage.
Avaliar os sistemas cognitivos de crianças e adolescentes com anemia falciforme provenientes de triagem neonatal e compará-las com controles sadios, ajustando-se os resultados para o nível socioeconômico.
MétodoA avaliação cognitiva foi feita com a escala de Wechsler WISC-III em 64 crianças e adolescentes com anemia falciforme e em 64 controles pareados por sexo e idade, sem a doença e sem comprometimento neurológico; o nível socioeconômico foi aferido pelo Critério Brasil.
ResultadosTodos os escores cognitivos foram inferiores no grupo de pacientes. As médias de QI Total, QI Verbal e QI de Execução foram respectivamente 90,95 para o grupo de pacientes e 113,97 para os controles (p<0,001); 91,41 para o grupo de pacientes e 112,31 para os controles (p<0,001); 92,34 para o grupo de pacientes e 113,38 para os controles (p<0,001). Os escores de velocidade de processamento, de resistência à distração e de organização perceptual foram, também, significativamente mais baixos nos pacientes. Detectou-se correlação direta e significativa entre o nível socioeconômico e os escores cognitivos. Em análise multivariada, para um mesmo nível socioeconômico, uma criança com anemia falciforme teve QI total, em média, 21,2 pontos mais baixo do que a média dos controles (p<0,001), indicou que a doença, ajustada para o efeito socioeconômico, é forte fator preditivo do QI total.
ConclusãoOs prejuízos cognitivos das crianças com anemia falciforme são intensos e se manifestam mesmo quando o efeito da doença é ajustado para o nível socioeconômico, o que, a nosso ver, requer abordagem preventiva precoce para tentar evitar tais prejuízos.
Sickle cell disease is a chronic clinical entity that, due to its incidence and biopsychosocial complexity, is considered to be one of the main public health problems of the present time. Worldwide, an estimated 275,000 children are born each year with sickle cell disease, 84% of them in Africa.1 In Brazil, the total number of patients is estimated at 30,000.2 In Minas Gerais, where the present study was carried out, the incidence in newborns is 1:1300.3
Hemoglobin S (HbS) is caused by a genetic mutation (GAG>GTG) at codon 6 of the HBB gene. In sickle cell anemia, the βS allele is in homozygous state. In the other clinical subtypes of sickle cell disease, double heterozygosis of the βS allele occurs with other alleles, such as βC, βD-Punjab, and β-thalassemia. HbS causes erythrocytes to acquire the sickle shape in a low oxygenation environment, causing vascular obstruction and hindering blood circulation. If the obstruction is severe, tissue hypoxia and ischemic infarctions with varying extent occur, depending on the size of the obstructed vessel and the existence of additional collateral circulation. Painful crises are the most common acute episodes, caused by vaso-occlusion. Chronic hemolysis and the recurrence of ischemic infarctions lead to lesions in several tissues and organs.4
Neurological events are regarded as the most complex clinical manifestations of the disease and are often associated with cognitive impairment. Cerebrovascular accident (CVA) was first reported in 1923, 13 years after the description of sickle cell anemia. However, this clinical manifestation did not draw attention until the 1970s, when studies of conventional brain angiography demonstrated the severity of cerebrovascular disease. Repeated (chronic) blood transfusions started being indicated as a prevention measure against recurrence after an initial episode of CVA.5
In addition to CVA and transient ischemic attack, which differs from the former because its focal neurological manifestations do not last for more than 24h, it is possible that patients with sickle cell anemia will develop so-called Silent Cerebral Infarction (SCI), which can be detected by Nuclear Magnetic Resonance (NMR) and is known as “silent” because evident clinical manifestations are not present. In most cases, these lesions manifest as poor school performance and/or cognitive impairment.6–10
In 1963, initial studies in the USA indicated that the intellectual development of children with sickle cell anemia would not change.11 From the 1980s onward, brain disease in individuals with sickle cell anemia started to be investigated more deeply. The studies then began to report neuropsychological damage and “delayed” academic performance in children with sickle cell anemia, when compared to others who did not have the disease.5
Therefore, children with sickle cell anemia usually show lower cognitive scores when compared to children without the disease. Worse global intellectual function has been reported in several studies, as well as deficits in specific areas such as executive function, selective attention, working memory, processing speed, vocabulary, abstract reasoning, and verbal comprehension. These deficits have been associated in several reports with the presence of SCI.12–17
The main objective of the present study was to evaluate the cognitive abilities of children and adolescents with sickle cell anemia, comparing them with matched controls without the disease and considering the possible effect of the families’ socioeconomic status.
MethodsThis was a cross-sectional study, with two groups of subjects: patients (Group 1) and controls (Group 2). In the first one, 63 patients with molecularly-confirmed sickle cell anemia (hemoglobin SS) and one with Sβ0 thalassemia (0% hemoglobin A and therefore hemoglobin genotype “SS” on hemoglobin electrophoresis) were assessed. Children who had a clinical history of ischemic CVA (overt stroke) or transient ischemic attack were excluded. The presence of SCI, detected by NMR in 20 of the 64 patients (31.2%), and abnormal flow velocity results in cerebral arteries by transcranial Doppler in 6 of 63 patients (9.5%) were not exclusion criteria. These tests, as well as clinical data on severity, laboratory results, and genetic tests, will be analyzed as possible risk factors for cognitive deficits in a complementary study that has yet to be published. Age ranged from 7 to 13 years; 37 individuals (57.8%) were females and 27 (42.2%) were males. All of them came from the State Neonatal Screening Program (Programa Estadual de Triagem Neonatal) and were being treated at Fundação Hemominas. They resided in the capital or in municipalities in the Metropolitan Region of Belo Horizonte, state of Minas Gerais, Brazil. Group 2 consisted of 64 controls (subjects without sickle cell disease), matched by gender and age to Group 1, who attended a municipal public school, located in Belo Horizonte.
The criterion of the Economic Classification of Brazil (Classificação Econômica do Brasil) – the criterion Brasil – was used as a tool for socioeconomic assessment. The questionnaire analyzes the ownership of household items (appliances, automobiles, number of bathrooms), number of employees, and the degree of education of the head of the family. The interviews for the socioeconomic status assessment were answered in person by the parents or guardians of the Group 1 subjects and carried out on the same day of the psychological tests. The interviews in Group 2 were answered by the parents or guardians of the subjects through phone calls.
The numerical results of the socioeconomic evaluation were transformed into categories and classified according to the recommendations of the utilized tool. The highest category (A1) ranged from 42 to 46 points; A2, 35–41; B1, 29–34; B2, 23–28; C1, 18–22; C2, 14–17; D, 8–13; and E, 0–7 points.
The psychometric evaluation was performed using the Wechsler Intelligence Scale for Children, 3rd edition (WISC-III). It consists of several subtests, evaluating distinct aspects of intelligence, and provides three measures: overall (full scale) IQ, performance IQ, and verbal IQ. Additionally, four factorial indexes are provided: Processing Speed (PS), Perceptual Organization (PO), Verbal Comprehension (VC), and Distraction Resistance (DR).
The WISC-III classifies the performance in the test based on the numerical results of the subject under evaluation. Children with a numerical score below 69 are classified as having intellectual disability; borderline, 70–79; low average, 80–89; average, 90–109; high average, 110–119; superior, 120–129; very superior, >130. In the statistical analysis, the intellectual retardation and borderline categories were grouped as “low”; low average, average, and high average as “average”; superior and very superior as “high.”
The statistical analysis was performed using SPSS software (IBM SPSS Statistics for Windows, Version 20.0. NY, USA). Quantitative results were expressed as the mean±Standard Deviation (SD), or median and interquartile range, when the distribution of values was non-Gaussian. Prevalence rates were expressed as percentages and 95% confidence intervals (CI). Comparisons between the means of the psychological test scores in different groups of children were performed with the t-test, using the statistical values corresponding to the homoscedastic (homogeneous variance) or heteroscedastic (heterogeneous variance) distribution, according to the Levene test. A bivariate linear regression equation was used to analyze the association between the psychological test scores (dependent variable) and the control/patient group adjusted for the socioeconomic status (independent variables). Statistical tests with alpha error probability ≤0.05 were considered statistically significant.
The study was carried out according to the principles of the Declaration of Helsinki. It was approved by the Research Ethics Committees of the two institutions involved in the study (ETIC 0005.0.203.000-10), and informed consents and terms of assent were signed by the patients’ parents/guardians, patients, and controls.
ResultsAs the groups were matched by gender and age, each group had 37 female participants and 27 males. The mean age in Group 1 was 10.8 years and in Group 2, 10.9 years.
Among the patients, 26 (40.6%) lived in the city capital of the state and 38 (59.3%) lived in other municipalities in the Metropolitan Region. Moreover, 60 children (93.7%) studied in public schools and only 4 (6.3%) in private schools. All 64 controls studied in a municipal public school, as previously mentioned.
The mean baseline hematological data of the patients were: total hemoglobin, 8.08g/dL (95% CI: 7.81–8.35); fetal hemoglobin, 14.70% (95% CI: 12.79–16.61); leukometry, 14.42×109/L (95% CI: 13.58–15.26); reticulocytes 14.64% (95% CI: 13.51–15.77).
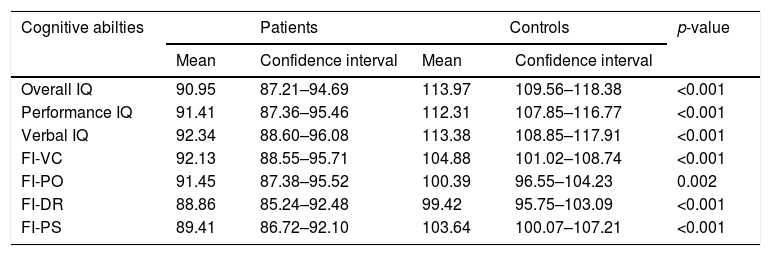
The WISC-III scores in the group of patients were lower in all IQ measurements when compared to the control group (Fig. 1). Table 1 summarizes the means and respective 95% CI of the IQ scores, as well as the PS, PO, VC, and DR factorial indexes for the two groups.
 scores, comparing 64 patients with sickle cell anemia with 64 controls. The difference between the two groups was statistically significant for the three quotients (p<0.001).")
Comparison of cognitive abilities between patients and controls.
| Cognitive abilties | Patients | Controls | p-value | ||
|---|---|---|---|---|---|
| Mean | Confidence interval | Mean | Confidence interval | ||
| Overall IQ | 90.95 | 87.21–94.69 | 113.97 | 109.56–118.38 | <0.001 |
| Performance IQ | 91.41 | 87.36–95.46 | 112.31 | 107.85–116.77 | <0.001 |
| Verbal IQ | 92.34 | 88.60–96.08 | 113.38 | 108.85–117.91 | <0.001 |
| FI-VC | 92.13 | 88.55–95.71 | 104.88 | 101.02–108.74 | <0.001 |
| FI-PO | 91.45 | 87.38–95.52 | 100.39 | 96.55–104.23 | 0.002 |
| FI-DR | 88.86 | 85.24–92.48 | 99.42 | 95.75–103.09 | <0.001 |
| FI-PS | 89.41 | 86.72–92.10 | 103.64 | 100.07–107.21 | <0.001 |
FI, factorial indexes; VC, verbal comprehension; PO, perceptual organization; DR, distraction resistance; PS, processing speed.
Regarding the socioeconomic status (SES), there was a statistically significant difference between the patient and control groups; mean of 18.33 (95% CI: 17.1–19.5), and 20.81 (95% CI: 19.7–21.9, p=0.004), respectively. Fig. 2 shows the statistically significant, direct correlation (p<0.001) between total IQ and SES of the 128 children (Pearson's correlation, r=0.31): the lower the Brasil score of a child, the lower is the overall IQ. This significant correlation was observed in a similar manner for all cognitive abilities analyzed. When the patients were analyzed separately (n=64), the correlation between overall IQ and SES was also statistically significant (p=0.04). For this reason, the impact of sickle cell disease on cognitive abilities was analyzed, adjusting the linear regression model for SES, as mentioned in the Methods section.
 and the socioeconomic status measured by the Brasil criterion, with scores ranging from zero to a maximum of 46 points (see Methods section); ■ patients, ○=controls.")
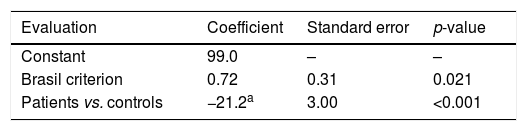
The data in Table 2 show that, for the same SES, children with sickle cell anemia had an overall IQ on average 21.2 points lower than the mean of the controls. Another example: children with sickle cell anemia (therefore, in the patient group) had the overall IQ increased by 0.72 for each increased point in the socioeconomic score. A third example, involving both variables: children with sickle cell anemia showing 10 points lower in the socioeconomic score than children from the control group had an overall IQ score that was 28.4 points lower (21.2+10×0.72).
Intelligence Quotient (IQ) scores of patient vs. controls, adjusted for the socioeconomic status measured by the Brasil criterion, obtained through bivariate linear regression.
| Evaluation | Coefficient | Standard error | p-value |
|---|---|---|---|
| Constant | 99.0 | – | – |
| Brasil criterion | 0.72 | 0.31 | 0.021 |
| Patients vs. controls | −21.2a | 3.00 | <0.001 |
Similarly, verbal IQ and performance IQ scores for children with sickle cell anemia, adjusted for SES, were 19.2 and 19.4 points lower when compared to control children (p<0.001 for both IQs). The same observation applies when factorial indexes were assessed, adjusted for the socioeconomic scores. The patients always showed lower scores than controls: the differences were 13.39 for processing speed and 10.92 for verbal comprehension (both with p<0.001); 9.66 for distraction resistance (p=0.001), and 7.5 points for perceptual organization (p=0.01).
DiscussionThe present study evaluated the cognitive abilities of children and adolescents with sickle cell anemia and compared this cognition in individuals without the disease. It was observed, as it had been hypothesized, that the patients had significantly lower cognitive scores than the controls in all assessed measures: overall, performance, and verbal IQ, and factorial indexes – verbal comprehension, distraction resistance, perceptual organization, and processing speed. This finding corroborates the many other studies that verified worse intellectual performance in patients with sickle cell disease; these studies have been submitted to a recently published meta-analysis.10
The Cooperative Study on Sickle Cell Disease (CSSCD) carried out in the United States was one of the first to prove that children and adolescents with sickle cell disease would be at greater risk of having cognitive function impairment.16
The aforementioned meta-analysis by Kawadler et al.10 reviewed 19 publications, reporting a decrease in overall IQ in patients with sickle cell disease. In these studies, on average, patients with a history of CVA showed a difference of ten IQ points when compared to patients with a history of SCI, who, in turn, had seven points less in their IQ score when compared to individuals without the disease. The results of the present study disclosed an even greater difference between patients and controls (23 points in overall IQ, 95% CI: 17.2–28.9), drawing attention to the magnitude of the damage the disease can cause.
In 2003, Steen et al.18 found lower scores in verbal and performance IQ in patients when compared to controls, whereas Noll et al.19 reported a difference only in verbal IQ. Differently from Noll's research, the present study, besides showing a difference in all cognitive evaluations, showed that there were also lower scores in the performance tasks.
Regarding the evaluations of more specific domains such as processing speed, distraction resistance, verbal comprehension, and perceptual organization, very significant differences were observed between the patient and the control groups in all cognitive abilities. Therefore, it was not possible to establish which cognitive function was the most affected one, as reported by Schatz et al.,15 who demonstrated that attention and executive function were the most impaired functions in patients with sickle cell disease.
The present study found, similarly to Schatz et al.,20 that patients with lower SES had equally lower overall IQ (p=0.04). As it is known, the families of individuals with sickle cell disease are mostly from the most disadvantaged socioeconomic groups. They are families that, historically, have had few social opportunities and, as a very likely consequence, belong to a low economic stratum.21 These families mostly show high social vulnerability, benefit from government social programs, and experience specific situations due to characteristics related to the disease itself, such as the fact that the mothers are unable to remain employed due to frequent hospitalizations of their children, frequent and recurrent blood transfusions, high rates of school dropout – most likely due to grade repetition caused by learning difficulties – and due to situations of racism and bullying at school. Moreover, significant family conflicts are more common in these families and have been associated with a lower IQ level in children with sickle cell anemia.22 Farber et al., in 1985, reported that the social conditions of the families of individuals with sickle cell disease were different from those of other families in the United States.11 This situation is similar for this population in Brazil.23
This global situation would explain the statistically significant difference of the mean socioeconomic score between the patients’ families and those of the controls. The ideal, from the methodological point of view, would have been for the control group to consist of students who attended the same school as the patient group. Such pairing would have the advantage of controlling the effect of the pedagogical quality of the school. However, this initiative would require logistics that the research team did not have. To minimize the issue, it was decided to evaluate children and adolescents who studied in a public school in the central region of the municipality of Belo Horizonte and that accepted students from more distant neighborhoods. Therefore, all the statistical analyses performed were adjusted for the socioeconomic effect, but it was not possible to control the influence of the pedagogical quality of the school.
The pathophysiological basis for IQ decline in children with sickle cell anemia is yet to be well understood. It is known that cognition, in general24 and specifically in children with sickle cell anemia, is affected by biological, socioeconomic, and cultural conditions.6,10,14,15,20 It is intuitive to consider the situation of chronic oxygen deficiency in the nervous tissue, due to anemia, as an underlying factor that would contribute primarily to the altered cognition. Significantly below-normal IQ scores have been associated, in several reports, with anemia and/or low oxygenation.8,10,14,18,20,25 The presence of SCI has been associated with low IQ scores in several reports,5–8,10,14,16–18,26 but there are studies that demonstrate that, even in the absence of SCI, children with sickle cell anemia have lower levels of cognition than those of controls.7,10,14,26,27 In a more recent study, it has been demonstrated that children with sickle cell anemia without SCI show evidence of alterations in the cerebral white matter morphometry.25
It is therefore necessary to develop strategies that can preventively minimize the risks of cognitive impairment, even in children whose symptoms would not be usually classified as severe. Based on this rationale for health prevention and promotion, the use of hydroxyurea might be recommended for all children with sickle cell anemia since an early age, as suggested by the main investigator of the Baby Hug study in a recent review article.28 It is imperative that clinical studies addressing the early use of hydroxyurea could measure, in addition to the usually controlled clinical and laboratory effects, the possible influence on the cognitive development of infants with sickle cell anemia.
The results of the present study also suggest that cognitive assessments should be included in the routine care of children with sickle cell anemia, since the eventual finding of an overall deficit or deficits in specific cognitive areas can guide school interventions, helping teachers and pedagogical teams regarding prevention and rehabilitation plans. This care has, therefore, the aim of maximizing the level of learning these children can achieve.6,15,26
Furthermore, as previously mentioned, the present study clearly corroborated the influence of SES on the cognition of students with sickle cell anemia. Therefore, it is necessary to establish strategies to improve the overall health status of these individuals, evidently including the environmental and social conditions.23 Hence, it is necessary to formulate public policies in the areas of education, health care, and social assistance that can improve access and ensure the permanence of these children and adolescents in educational institutions.
In conclusion, the present study showed that cognitive impairment in children with sickle cell anemia, even considering concurrent socioeconomic factors, is a significant finding, requiring additional studies that can address the pathophysiological aspects, aiming to allow the adoption of preventive measures.
FundingBrazilian Ministry of Health, CNPq, Nupad–UFMG.
Conflicts of interestThe authors declare no conflicts of interest.
To CNPq, The Brazilian Ministry of Health, and Nupad for financial support to the project. To the colleagues of Fundação Hemominas and Nupad for their logistic support. To the undergraduate students Luana Evelyn Abreu Oliveira Nery and Ana Clara Rocha Franco for the help in applying the WISC-III. To the children and families for their consent to carry out the study and publish the results.
Please cite this article as: Castro IP, Viana MB. Cognitive profile of children with sickle cell anemia compared to healthy controls. J Pediatr (Rio J). 2019;95:451–7.
Study conducted at Universidade Federal de Minas Gerais (UFMG), Faculdade de Medicina, Departamento de Pediatria; and Universidade Federal de Minas Gerais (UFMG), Faculdade de Medicina, Núcleo de Ações e Pesquisa em Apoio Diagnóstico (Nupad), Fundação Hemominas, Belo Horizonte, MG, Brazil.




