
Literature review of new genes related to osteogenesis imperfecta (OI) and update of its classification.
SourcesLiterature review in the PubMed and OMIM databases, followed by selection of relevant references.
Summary of the findingsIn 1979, Sillence et al. developed a classification of OI subtypes based on clinical features and disease severity: OI type I, mild, common, with blue sclera; OI type II, perinatal lethal form; OI type III, severe and progressively deforming, with normal sclera; and OI type IV, moderate severity with normal sclera. Approximately 90% of individuals with OI are heterozygous for mutations in the COL1A1 and COL1A2 genes, with dominant pattern of inheritance or sporadic mutations. After 2006, mutations were identified in the CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, WNT1, BMP1, and TMEM38B genes, associated with recessive OI and mutation in the IFITM5 gene associated with dominant OI. Mutations in PLS3 were recently identified in families with osteoporosis and fractures, with X‐linked inheritance pattern. In addition to the genetic complexity of the molecular basis of OI, extensive phenotypic variability resulting from individual loci has also been documented.
ConclusionsConsidering the discovery of new genes and limited genotype‐phenotype correlation, the use of next‐generation sequencing tools has become useful in molecular studies of OI cases. The recommendation of the Nosology Group of the International Society of Skeletal Dysplasias is to maintain the classification of Sillence as the prototypical form, universally accepted to classify the degree of severity in OI, while maintaining it free from direct molecular reference.
Revisão da literatura sobre novos genes relacionados à osteogênese imperfeita (OI) e atualização da sua classificação.
Fonte dos dadosRevisão nas bases de dados do PUBMED e OMIM com seleção de referências relevantes.
Síntese dos dadosSillence et al., em 1979, desenvolveram uma classificação dos subtipos de OI baseada em características clínicas e gravidade da doença: OI tipo I, forma leve, comum, com escleras azuladas; OI tipo II, forma perinatal letal; OI tipo III, forma grave e progressivamente deformante com esclera normal; e OI tipo IV, forma de gravidade moderada com esclera normal. Cerca de 90% dos indivíduos com OI são heterozigotos para mutações em COL1A1 e COL1A2, com padrão de herança dominante ou esporádico. A partir de 2006 foram identificadas mutações nos genes CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, WNT1, BMP1 e TMEM38B associadas à OI recessiva e mutação em IFITM5 associada à OI dominante. Mutações em PLS3 foram identificadas recentemente em famílias com osteoporose e fraturas, com padrão de herança ligado ao X. Além da complexidade genética das bases moleculares das OI, extensa variabilidade fenotípica resultante de loci individuais também tem sido documentada.
ConclusõesFace à descoberta de novos genes e à correlação genótipo‐fenótipo limitada, o uso de ferramentas de sequenciamento de nova geração torna‐se útil no estudo molecular de casos de OI. A recomendação do Grupo de Nosologia da Sociedade Internacional de Displasias Esqueléticas é manter a classificação de Sillence como a forma prototípica e universalmente aceita para classificar o grau de gravidade na OI, e libertá‐la de referência molecular direta.
Osteogênese imperfeita (OI) é um grupo de doenças clinica e geneticamente heterogêneo, caracterizado por suscetibilidade a fraturas ósseas com gravidade variável e defeitos presumidos ou comprovados na biossíntese de colágeno tipo I. Outras manifestações são dentinogênese imperfeita, escleras azuis, baixa estatura, e perda auditiva na idade adulta. As manifestações clínicas variam em um continuum que vai desde casos graves, com letalidade perinatal, até indivíduos assintomáticos, com predisposição leve a fraturas, estatura e vida normais.1
Em, a incidência dos vários tipos de OI é de cerca de 1 em 15.000‐20.000 nascimentos, a maioria de herança autossômica dominante por mutação em COL1A1 ou COL1A2, que codificam as cadeias α1(I) e α2(I) de colágeno tipo I.1
O colágeno tipo I, principal proteína estrutural da matrix extracelular dos ossos, pele e tendões, é composto de duas pró‐cadeias α‐1 e uma pró‐cadeia α‐2, que se entrelaçam formando tripla hélice rígida. Cada cadeia α contém pró‐peptídeos terminais nas extremidades C‐terminal (carboxi) e N‐terminal (amino) e um domínio central composto de 338 repetições de Gly‐X‐Y, onde o X e o Y excluem cisteína e triptofano, e frequentemente são, respectivamente, prolina e hidroxiprolina. A glicina, por ser o menor aminoácido, é o único resíduo capaz de ocupar a posição axial da tripla hélice, de modo que qualquer alteração em um resíduo de glicina acarretará desorganização da estrutura helicoidal.2,3
As mutações em COL1A1 e COL1A2 alteram a estrutura ou a quantidade de colágeno tipo I e causam um fenótipo esquelético que varia de subclínico a letal.1 Estes pacientes apresentam anomalias qualitativas e quantitativas no colágeno tipo I devido ao efeito dominante negativo da mutação, já que as pró‐cadeias α mutantes são incorporadas nas moléculas de pró‐colágeno tipo I, que contêm também pró‐cadeias α normais. Como regra, quando há substituição da glicina na cadeia α1, e o fenótipo vai depender da posição da substituição: substituições C‐terminais causam um fenótipo grave da doença e as substituições N‐terminais, fenótipos mais leves.4,5 Resíduos com cadeias laterais grandes ou carregados são altamente desorganizadores da estrutura tripla, não importando onde estejam localizados. Diferentes fenótipos têm sido encontrados com a mesma mutação.6
Em consórcio realizado em 2007 para estudo de mutações causadoras de OI nos genes de colágeno 1, foram identificadas 1.832 mutações independentes, sendo 682 resultado de substituição de resíduos de glicina no domínio da tripla hélice da proteína codificada e 150 de locais de sítio de splice.6
Com base em achados clínicos, achados radiográficos do esqueleto, modo de herança e análises genéticas moleculares, novas OI vêm sendo identificadas a partir de 2006, por meio de sequenciamento do exoma. O presente trabalho teve por objetivo rever a classificação das OI e atualizar os novos genes relacionados. Foram utilizadas as bases de dados do PUBMED e do Mendelian Inheritance in Man (MIM).7
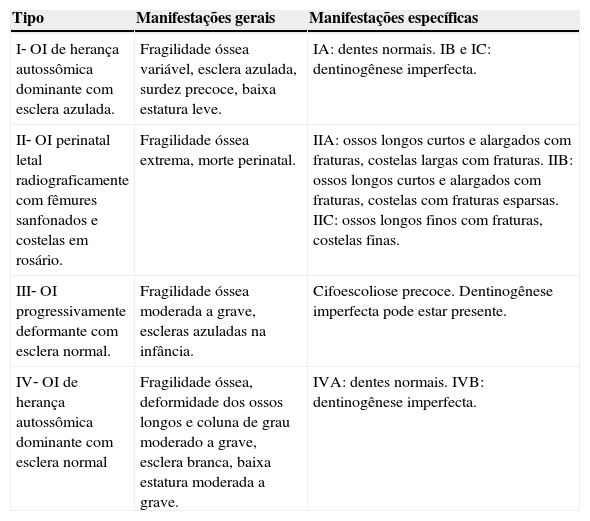
Classificação de SillenceDevido à variabilidade fenotípica considerável, Sillence et al.8,9 desenvolveram uma classificação dos subtipos OI baseada em características clínicas e gravidade da doença (tabela 1): OI tipo I, forma leve, comum, com escleras azuladas; OI tipo II, forma perinatal letal; OI tipo III, forma grave e progressivamente deformante, com esclera normal; e OI tipo IV, forma de gravidade moderada, com esclera normal. A classificação de Sillence vem sendo repetidamente revista em momentos de identificação de novos genes causadores da OI.
Classificação de OIa
| Tipo | Manifestações gerais | Manifestações específicas |
|---|---|---|
| I‐ OI de herança autossômica dominante com esclera azulada. | Fragilidade óssea variável, esclera azulada, surdez precoce, baixa estatura leve. | IA: dentes normais. IB e IC: dentinogênese imperfecta. |
| II‐ OI perinatal letal radiograficamente com fêmures sanfonados e costelas em rosário. | Fragilidade óssea extrema, morte perinatal. | IIA: ossos longos curtos e alargados com fraturas, costelas largas com fraturas. IIB: ossos longos curtos e alargados com fraturas, costelas com fraturas esparsas. IIC: ossos longos finos com fraturas, costelas finas. |
| III‐ OI progressivamente deformante com esclera normal. | Fragilidade óssea moderada a grave, escleras azuladas na infância. | Cifoescoliose precoce. Dentinogênese imperfecta pode estar presente. |
| IV‐ OI de herança autossômica dominante com esclera normal | Fragilidade óssea, deformidade dos ossos longos e coluna de grau moderado a grave, esclera branca, baixa estatura moderada a grave. | IVA: dentes normais. IVB: dentinogênese imperfecta. |
A classificação genética molecular de OI tem se revelado muito heterogênea, com diversos padrões de herança e ampla variabilidade de gravidade clínica. 10
Glorieux et al.11 descreveram uma forma autossômica dominante de OI, similar à OI tipo IV de Sillence, mas com características clínica, histológica e molecular distintas. Não foi encontrada mutação em COL1A1 e COL1A2, sendo então nomeada pelos autores de OI tipo V (MIM #610967). Cerca de 65% dos indivíduos afetados desenvolvem calos hiperplásicos após fraturas ou intervenções cirúrgicas, considerada uma característica patognomônica.12 Somente em 2012 foram identificadas mutações no IFITM5 em pacientes com OI tipo V, gene que codifica a proteína 5 transmembrana interferon‐induzida, por sequenciamento de todo o exoma.12–14 A proteína codificada tem papel na mineralização precoce, mas seu mecanismo é desconhecido.10
Em 2006, mutação no gene CRTAP foi identificada como primeira causa genética de OI recessiva letal.15 A partir de, mutações em novos genes que causam OI recessiva têm sido identificadas por sequenciamento do exoma, como FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP, BMP1 e TMEM38B. Cada um destes genes recebeu um número de tipo de OI na base de dados MIM, dando sequência aos números de classificação de Sillence.
A OI chamada tipo VI (MIM #613982) é uma forma autossômica recessiva da doença, que pode ser causada por mutação homozigótica no gene SERPINF1 em 17p13.3, com defeito de mineralização.14 Pela classificação de Sillence, o fenótipo é compatível com o tipo IV ou tipo III.16,17
OI tipo VII (MIM #610682) é uma forma autossômica recessiva letal de OI causada por mutação no gene CRTAP em homozigose ou heterozigose composta no cromossomo 3p22. É responsável por 2 a 3% dos casos de OI letal.15
Cabral et al.18 descreveram uma forma de OI autossômica recessiva, denominada OI tipo VIII (MIM #610915), que se caracteriza por esclera branca, grave deficiência de crescimento, mineralização esquelética muito deficiente e metáfises bulbosas. Esta forma é causada por mutação no gene que codifica leprecan (LEPRE1), em 1p34.2, associada à OI grave ou letal.
OI tipo IX (MIM #259440) é uma forma autossômica recessiva de OI correspondente aos tipos clinicamente graves II / III da classificação de Sillence.19 Não há relato de dentinogênese imperfeita. Ela pode ser causada por mutação homozigótica do gene PPIB em 15q22.31.
OI tipo X (MIM #613848) é uma forma autossômica recessiva, que pode ser causada por uma mutação homozigótica do gene SERPINH1 no cromossomo 11q13.5. É caracterizada por deformidades ósseas e fraturas múltiplas, osteopenia generalizada, dentinogênese imperfeita e esclera azulada. SERPINH1 codifica uma proteína de colágeno de ligação que funciona como uma chaperona no retículo endoplasmático, motivo pelo qual os indivíduos com mutação neste gene apresentam células que não produzem colágeno tipo I supermodificado.20
OI tipo XI (MIM #610968) é uma forma autossômica recessiva causada por uma mutação homozigótica do gene FKBP10 em 17q21, também relacionada a defeito de chaperona.1 Pacientes com OI tipo XI apresentam deformação progressiva grave e podem ter contraturas articulares. Os pacientes não apresentam dentinogênese imperfeita.21–23
OI tipo XII (MIM #613849) é uma forma autossômica recessiva, que pode ser causada por mutação no gene SP7 em 12q13.13. Clinicamente é caracterizada por fraturas recorrentes, deformações ósseas leves, osteoporose generalizada, atraso da erupção dos dentes, ausência de dentinogênese imperfeita, audição normal e esclerótica branca.24
OI tipo XIII (MIM #614856) foi descrito por mutação homozigótica no gene BMP1 no cromossomo 8p21.25,26
Shaheen et al.27 descreveram a OI tipo XIV (MIM #615066), uma forma autossômica recessiva caracterizada por graus variáveis de gravidade de múltiplas fraturas e osteopenia, com dentes, esclera e audição normais. Fraturas ocorrem no pré‐natal ou por volta dos seis anos de idade. É causada por mutação homozigótica no gene TMEM38B no cromossomo 9q31.
OI tipo XV (MIM #615220) foi nomeada a partir da identificação de mutações em WNT1.28–30 Keupp et al.30 reportaram que os alelos hipofuncionais de WNT1 causam fenótipos com baixa massa óssea em humanos. Identificaram que mutações no gene herdadas de forma recessiva levam a fenótipos de gravidades variáveis, variando de formas moderadas a progressivamente deformantes, podendo, ocasionalmente, levar à morte infantil precoce. Detectaram também famílias com padrão autossômico dominante de osteoporose precoce apresentando mutação heterozigótica em WNT1.
As formas recessivas de OI com fenótipos letais a moderados são causadas por defeitos em genes cujos produtos interagem com o colágeno tipo I. A maioria dos casos recessivos tem mutações nulas em genes que codificam proteínas envolvidas na prolil 3‐hidroxilação do colágeno (CRTAP, LEPRE1 e PPIB) ou as responsáveis pela correta dobragem helical (FKBP10 e SERPINH1). Os tipos VII, VIII e IX são causados por defeitos de 3‐hidroxilação.1 A correlação genótipo‐fenótipo nas formas recessivas tem sido sugerida.31
Em 2013 mutações em PLS3 foram identificadas em famílias com osteoporose e fraturas se manifestando na infância, de herança ligada ao X.32
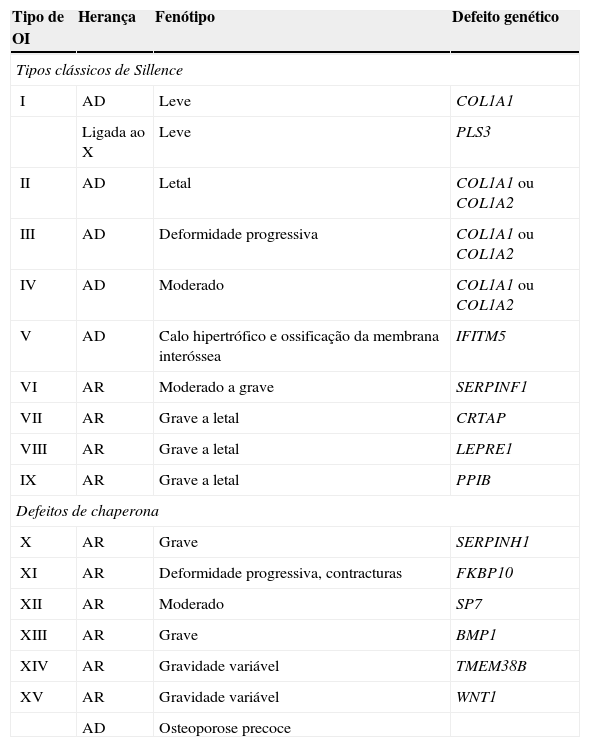
A tabela 2 resume a classificação baseada em genes envolvidos.
Nosologia da OIa
| Tipo de OI | Herança | Fenótipo | Defeito genético |
|---|---|---|---|
| Tipos clássicos de Sillence | |||
| I | AD | Leve | COL1A1 |
| Ligada ao X | Leve | PLS3 | |
| II | AD | Letal | COL1A1 ou COL1A2 |
| III | AD | Deformidade progressiva | COL1A1 ou COL1A2 |
| IV | AD | Moderado | COL1A1 ou COL1A2 |
| V | AD | Calo hipertrófico e ossificação da membrana interóssea | IFITM5 |
| VI | AR | Moderado a grave | SERPINF1 |
| VII | AR | Grave a letal | CRTAP |
| VIII | AR | Grave a letal | LEPRE1 |
| IX | AR | Grave a letal | PPIB |
| Defeitos de chaperona | |||
| X | AR | Grave | SERPINH1 |
| XI | AR | Deformidade progressiva, contracturas | FKBP10 |
| XII | AR | Moderado | SP7 |
| XIII | AR | Grave | BMP1 |
| XIV | AR | Gravidade variável | TMEM38B |
| XV | AR | Gravidade variável | WNT1 |
| AD | Osteoporose precoce | ||
AD, autossômica dominante; AR, autossômica recessiva.
Em 2010, van Dijk et al.33 propuseram uma classificação revisada das OI, mencionando o gene causador e o quadro clínico indicado apenas para os tipos de I a VI. Os tipos VII e VIII foram excluídos, uma vez que estes haviam sido adicionados por critérios genéticos, embora seus achados clínicos e radiológicos fossem indistinguíveis dos tipos II a IV. A classificação proposta deixa espaço para novos genes descobertos como causa de OI até que a extensão da heterogeneidade seja conhecida.34
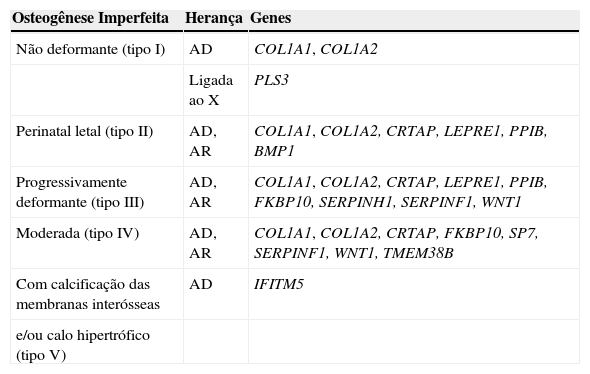
Classificação das OI pela Sociedade Internacional de Displasias EsqueléticasPela alta complexidade genética das bases moleculares das OI e extensa variabilidade fenotípica resultante de loci individuais descrita nos últimos anos, parecia insustentável manter correlações entre os tipos de Sillence e sua base molecular. Porém, a proliferação dos tipos de OI para refletir cada gene separadamente, defendida por alguns, se tornou mais confusa do que útil na prática clínica. Por esses motivos, o Grupo de Nosologia da Sociedade Internacional de Displasias Esqueléticas, reunido em 2009, recomendou manter a classificação de Sillence como a forma prototípica e universalmente aceita para classificar o grau de gravidade na OI e libertá‐la de referência molecular direta.35 Assim, como listadas na tabela 3, as OI foram agrupadas em cinco categorias clínicas, e os vários genes que podem causar OI foram listados separadamente. Acrescentamos à tabela original os genes IFITM5, SERPINF1, BMP1, WNT1, TMEM38B e PLS3, descobertos depois da sua publicação.
Classificação das OI pela Sociedade Internacional de Displasias Esqueléticasa com acréscimo de genes recentemente descobertos
| Osteogênese Imperfeita | Herança | Genes |
|---|---|---|
| Não deformante (tipo I) | AD | COL1A1, COL1A2 |
| Ligada ao X | PLS3 | |
| Perinatal letal (tipo II) | AD, AR | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, BMP1 |
| Progressivamente deformante (tipo III) | AD, AR | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1, SERPINF1, WNT1 |
| Moderada (tipo IV) | AD, AR | COL1A1, COL1A2, CRTAP, FKBP10, SP7, SERPINF1, WNT1, TMEM38B |
| Com calcificação das membranas interósseas | AD | IFITM5 |
| e/ou calo hipertrófico (tipo V) |
AD, autossômica dominante; AR, autossômica recessiva.
Na prática, apesar da complexa variabilidade genotípica da OI evidenciada nos últimos anos, seus fenótipos ainda se enquadram na classificação de Sillence. A investigação genotípica deve ser indicada especialmente nos casos em que sugiram herança autossômica recessiva, a fim de aconselhamento genético. O estudo molecular deve ser feito por meio de sequenciamento de Sanger dos diversos novos genes ou por sequenciamento de nova geração. O sequenciamento do exoma tem utilidade quando não há um painel de genes disponível, ou quando não se conhecem os genes envolvidos.
FinanciamentoCNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico).
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) por ter proporcionado, em 2013, a bolsa de pós‐doutorado de Eugênia Ribeiro Valadares no setor de genética da Clínica Pediátrica da Universidade de Freiburg, Alemanha, para desenvolver o projeto “Investigação de osteogênese imperfeita pela análise dos genes conhecidos e novos genes candidatos em pacientes brasileiros e alemães”, sob supervisão do Prof. Dr. Bernhard Zabel, do Dr. Pablo Villavicencio Lorini e do Dr. Ekkehart Lausch, pessoas do nosso maior apreço.
Como citar este artigo: Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics and osteogenesis imperfecta classification? J Pediatr (Rio J). 2014;90:536–41.




