

Literature review of new genes related to osteogenesis imperfecta (OI) and update of its classification.
SourcesLiterature review in the PubMed and OMIM databases, followed by selection of relevant references.
Summary of the findingsIn 1979, Sillence et al. developed a classification of OI subtypes based on clinical features and disease severity: OI type I, mild, common, with blue sclera; OI type II, perinatal lethal form; OI type III, severe and progressively deforming, with normal sclera; and OI type IV, moderate severity with normal sclera. Approximately 90% of individuals with OI are heterozygous for mutations in the COL1A1 and COL1A2 genes, with dominant pattern of inheritance or sporadic mutations. After 2006, mutations were identified in the CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, WNT1, BMP1, and TMEM38B genes, associated with recessive OI and mutation in the IFITM5 gene associated with dominant OI. Mutations in PLS3 were recently identified in families with osteoporosis and fractures, with X-linked inheritance pattern. In addition to the genetic complexity of the molecular basis of OI, extensive phenotypic variability resulting from individual loci has also been documented.
ConclusionsConsidering the discovery of new genes and limited genotype-phenotype correlation, the use of next-generation sequencing tools has become useful in molecular studies of OI cases. The recommendation of the Nosology Group of the International Society of Skeletal Dysplasias is to maintain the classification of Sillence as the prototypical form, universally accepted to classify the degree of severity in OI, while maintaining it free from direct molecular reference.
Revisão da literatura sobre novos genes relacionados à osteogênese imperfeita (OI) e atualização da sua classificação.
Fonte dos dadosRevisão nas bases de dados do PUBMED e OMIM com seleção de referências relevantes.
Síntese dos dadosSillence et al., em 1979, desenvolveram uma classificação dos subtipos de OI baseada em características clínicas e gravidade da doença: OI tipo I, forma leve, comum, com escleras azuladas; OI tipo II, forma perinatal letal; OI tipo III, forma grave e progressivamente deformante com esclera normal; e OI tipo IV, forma de gravidade moderada com esclera normal. Cerca de 90% dos indivíduos com OI são heterozigotos para mutações em COL1A1 e COL1A2, com padrão de herança dominante ou esporádico. A partir de 2006 foram identificadas mutações nos genes CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, WNT1, BMP1 e TMEM38B associadas à OI recessiva e mutação em IFITM5 associada à OI dominante. Mutações em PLS3 foram identificadas recentemente em famílias com osteoporose e fraturas, com padrão de herança ligado ao X. Além da complexidade genética das bases moleculares das OI, extensa variabilidade fenotípica resultante de loci individuais também tem sido documentada.
ConclusõesFace à descoberta de novos genes e à correlação genótipo-fenótipo limitada, o uso de ferramentas de sequenciamento de nova geração torna-se útil no estudo molecular de casos de OI. A recomendação do Grupo de Nosologia da Sociedade Internacional de Displasias Esqueléticas é manter a classificação de Sillence como a forma prototípica e universalmente aceita para classificar o grau de gravidade na OI, e libertá-la de referência molecular direta.
Osteogenesis imperfecta (OI) is a group of clinically and genetically heterogeneous diseases characterized by susceptibility to bone fractures, with variable degree of severity and presumed or proven defects in collagen type I biosynthesis. Other manifestations include dentinogenesis imperfecta, blue sclerae, and short stature, as well as hearing loss in adulthood. Clinical manifestations range from severe cases with perinatal lethality to asymptomatic individuals with mild predisposition to fractures, normal stature, and normal life.1
Overall, the incidence of the different types of OI is approximately 1 in 15,000-20,000 births and most cases are due to autosomal dominant inheritance with mutations in COL1A1 or COL1A2 genes, which encode the α1 (I) and α2 (I) chains of type I collagen.1
Type I collagen, the main structural protein of the extracellular matrix of bone, skin, and tendons, consists of two pro-α-1 chains and one pro-α-2 chain that interweave, forming a rigid triple helix. Each α chain contains N-(amino) and C-(carboxy) terminal propeptides and a central domain consisting of 338 repeats of Gly-XY, where X and Y exclude cysteine and tryptophan, and which often are, respectively, proline and hydroxyproline. Glycine, as the smallest amino acid, is the only residue that can occupy the axial position of the triple helix, so that any change in a glycine residue will result in the disruption of the helical structure.2,3
Mutations in COL1A1 and COL1A2 genes alter the structure or the amount of type I collagen, resulting in a skeletal phenotype that ranges from subclinical to lethal.1
These patients exhibit qualitative and quantitative abnormalities in type I collagen due to the dominant negative effect of the mutation, as the mutant pro-α chains are incorporated into the type I procollagen molecules that also contain normal pro-α chains. As a rule, when there is substitution of glycine in the α1 chain, the phenotype will depend on the position of the substitution: C-terminal substitutions result in severe disease phenotype, and N-terminal substitutions yield milder phenotypes.4,5 Residues with large lateral chains or charged residues are highly disruptive of the triple structure, regardless of where they are located. Different phenotypes have been found with the same mutation.6
In a consortium created in 2007 to study OI-causing mutations in type I collagen genes, 1,832 independent mutations were identified; 682 resulted in the substitution of glycine residues in the triple helix domain of the encoded protein, and 150 in splice sites.6
Based on clinical, radiographic, and skeletal findings, mode of inheritance, and molecular genetic analyses, new OI types have been identified since 2006 through exome sequencing. The present study aimed to review the classification of OI and to update new related genes. The PubMed and Online Mendelian Inheritance in Man (OMIM) databases were used.7
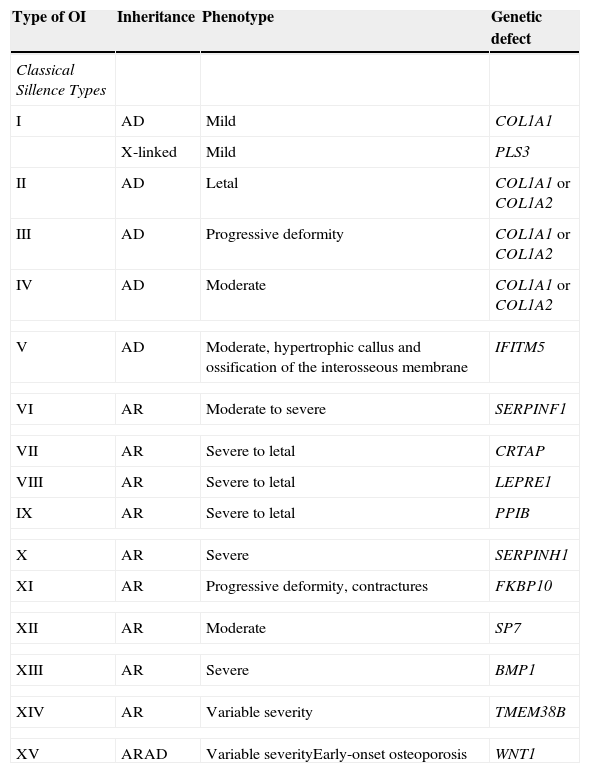
Sillence ClassificationDue to considerable phenotypic variability, Sillence et al.8,9 developed a classification of OI subtypes based on clinical features and disease severity (Table 1): OI type I, mild, common, with blue sclera; OI type II, perinatal lethal form; OI type III, severe and progressively deforming, with normal sclera; and OI type IV, moderate severity with normal sclera. The classification of Sillence has been repeatedly revised when new causative genes for OI are identified.
Classification of OI.a
| Type | General manifestations | Specific manifestations |
|---|---|---|
| I- Autosomal dominant inheritance with blue sclera. | Variable bone fragility, blue sclera, early deafness, mild stunting. | I-A: normal teeth. |
| I-B and I-C: dentinogenesis imperfecta. | ||
| II- Perinatal lethal form, radiographically characterized by crumpled femora and beaded ribs. | Extreme bone fragility, perinatal death. | II-A: short and widened long bones with fractures, wide ribs with fractures. |
| II-B: short and widened long bones with fractures, ribs with sparse fractures. | ||
| II-C: thin long bones with fractures, thin ribs. | ||
| III- Progressively deforming, with normal sclera. | Moderate to severe bone fragility, blue sclera in infancy. | Early-onset kyphoscoliosis. |
| Dentinogenesis imperfecta may be present. | ||
| IV- Autosomal dominant inheritance with normal sclera. | Bone fragility, moderate to severe deformity of the long bones and spinal column, white sclera, moderate to severe stunting. | IV-A: normal teeth. |
| IV-B: dentinogenesis imperfecta. |
The molecular genetic classification of OI has shown to be very heterogeneous, with different patterns of inheritance and wide variability of clinical severity.10
Glorieux et al.11 described an autosomal dominant form of OI, similar to OI Sillence type IV, but with distinct clinical, histological, and molecular characteristics. No mutations were found in COL1A1 and COL1A2 and, therefore, it was called OI type V (OMIM #610967) by the authors. Approximately 65% of affected individuals develop hyperplastic callus after fractures or surgical interventions, considered a pathognomonic characteristic.12 Only in 2012 were IFITM5 mutations identified in patients with type V OI, the gene encoding interferon-induced transmembrane protein 5, by sequencing of the entire exome.12–14 The encoded protein has a role in early mineralization, but its mechanism remains unknown.10
In 2006, a CRTAP gene mutation was identified as the first genetic cause of lethal recessive OI.15 Since then, new mutations in genes that cause recessive OI have been identified by exome sequencing, such as FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP, BMP1, and TMEM38B. Each of these genes received an OI type number in the OMIM database, following the sequence numbers of the Sillence classification.
OI type VI (OMIM #613982) is an autosomal recessive form of the disease that can be caused by a homozygous mutation in the gene SERPINF1 in chromosome 17p13.3, causing a mineralization defect.14 According to the classification of Sillence et al., the phenotype is compatible with type IV or type III.16,17
OI type VII (MIM #610682) is a lethal autosomal recessive form of OI, caused by a mutation in CRTAP gene in homozygosity or compound heterozygosity in chromosome 3p22. It accounts for 2% to 3% of cases of lethal OI.15
Cabral et al.18 described a form of autosomal recessive OI, called OI type VIII (OMIM #610915), which is characterized by white sclera, severe growth impairment, very poor skeletal mineralization, and bulbous metaphyses. This form is caused by mutations in the gene encoding leprecan (LEPRE1) in chromosome 1p34.2, associated with severe or lethal OI.
OI type IX (OMIM #259440) is an autosomal recessive form of OI corresponding to clinically severe types II/III of the Sillence classification.19 There are no reports of dentinogenesis imperfecta. It can be caused by a homozygous mutation in the PPIB gene in chromosome 15q22.31.
OI type X (OMIM #613848) is an autosomal recessive form of the disease that can be caused by a homozygous mutation in the gene SERPINH1 in chromosome 11q13.5. It is characterized by bone deformities and multiple fractures, generalized osteopenia, dentinogenesis imperfecta, and blue sclera. SERPINH1 encodes a collagen-binding protein that acts as a chaperone in the endoplasmic reticulum and thus, individuals with mutations in this gene have cells that do not produce overmodified type I collagen.20
OI type XI (OMIM #610968) is an autosomal recessive form of the disease caused by a homozygous mutation in the FKBP10 gene in chromosome 17q21, also related to a chaperone defect.1 Patients with type OI type XI have severe progressive deformation and may have joint contractures. Patients do not have dentinogenesis imperfecta.21–23
OI type XII (OMIM #613849) is an autosomal recessive form, which can be caused by mutation in the SP7 gene in chromosome 12q13.13. It is clinically characterized by recurrent fractures, mild bone deformities, generalized osteoporosis, delayed eruption of teeth, absence of dentinogenesis imperfecta, normal hearing, and white sclera.24
OI type XIII (OMIM #614856) was described as caused by a homozygous mutation in the gene BMP1 in chromosome 8p21.25,26
Shaheen et al.27 described OI type XIV (OMIM # 615066), an autosomal recessive form characterized by varying degrees of severity with multiple fractures and osteopenia, with normal dentition, sclera, and hearing. Fractures occur prenatally or at approximately 6 years of age. It is caused by a homozygous mutation in the gene TMEM38B in chromosome 9q31.
OI type XV (OMIM #615220) has been designated based on the identification of mutations in WNT1.28–30 Keupp et al.30 reported that WNT1 hypofunctional alleles result in phenotypes with low bone mass in humans. They verified that mutations in the recessive inherited gene lead to phenotypes of varying severity, ranging from mild to progressively deforming, which can occasionally lead to early infant death. They also detected families that had early osteoporosis with the autosomal dominant pattern of inheritance, with a heterozygous mutation in WNT1.
The recessive forms of OI with moderate to lethal phenotypes are caused by defects in genes whose products interact with collagen type I. Most recessive cases have null mutations in genes encoding proteins involved in prolyl 3-hydroxylation of collagen (CRTAP, LEPRE1, and PPIB), or in those responsible for the correct helical folding (FKBP10 and SERPINH1). Types VII, VIII, and IX are caused by defects in 3-hydroxylation.1 The genotype-phenotype correlation in recessive forms has been suggested.31
In 2013, PLS3 mutations were identified in families with osteoporosis and fractures manifesting in childhood, with an X-linked pattern of inheritance.32
Table 2 summarizes the classification based on the involved genes.
Expanded classification of OI.a
| Type of OI | Inheritance | Phenotype | Genetic defect |
|---|---|---|---|
| Classical Sillence Types | |||
| I | AD | Mild | COL1A1 |
| X-linked | Mild | PLS3 | |
| II | AD | Letal | COL1A1 or COL1A2 |
| III | AD | Progressive deformity | COL1A1 or COL1A2 |
| IV | AD | Moderate | COL1A1 or COL1A2 |
| V | AD | Moderate, hypertrophic callus and ossification of the interosseous membrane | IFITM5 |
| VI | AR | Moderate to severe | SERPINF1 |
| VII | AR | Severe to letal | CRTAP |
| VIII | AR | Severe to letal | LEPRE1 |
| IX | AR | Severe to letal | PPIB |
| X | AR | Severe | SERPINH1 |
| XI | AR | Progressive deformity, contractures | FKBP10 |
| XII | AR | Moderate | SP7 |
| XIII | AR | Severe | BMP1 |
| XIV | AR | Variable severity | TMEM38B |
| XV | ARAD | Variable severityEarly-onset osteoporosis | WNT1 |
AD, autosomal dominant; AR, autosomal recessive.
In 2010, van Dijk et al.33 proposed a revised classification of OI, mentioning the causative gene and the corresponding clinical picture only for types I to VI. Types VII and VIII were excluded, as those types were added by genetic criteria, although their clinical and radiological findings were indistinguishable from those in types II to IV. The proposed classification leaves room for new genes discovered as the cause of OI until the full extent of heterogeneity is known.34
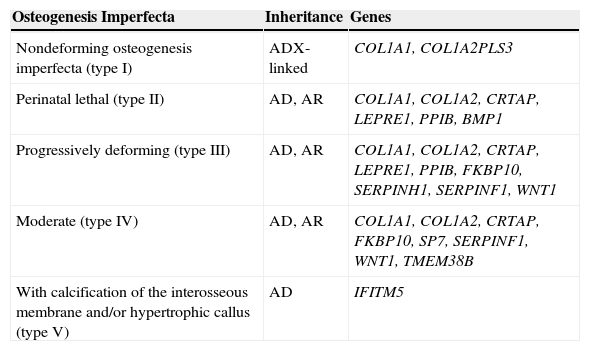
OI Classification by the International Society of Skeletal DysplasiasDue to the high genetic complexity of the molecular basis of OI and the extensive phenotypic variability resulting from individual loci described in recent years, it seemed impossible to maintain correlations between Sillence types and their molecular basis. But the proliferation of OI types to reflect each gene separately, supported by some, has become more confusing than useful in clinical practice. For these reasons, in 2009 the Nosology Group of the International Society of Skeletal Dysplasias recommended maintaining the classification of Sillence as the prototypical and universally accepted form to classify the degree of OI severity, and freeing it from direct molecular reference.35 Thus, as shown in Table 3, OI was grouped into five clinical categories, and the several genes that can cause OI were listed separately. In the present study, the genes IFITM5, SERPINF1, BMP1, WNT1, TMEM38B, and PLS3 were added to the original table, as they were discovered after its publication.
OI classification according to the International Society of Skeletal Dysplasiasa with addition of newly discovered genes.
| Osteogenesis Imperfecta | Inheritance | Genes |
|---|---|---|
| Nondeforming osteogenesis imperfecta (type I) | ADX-linked | COL1A1, COL1A2PLS3 |
| Perinatal lethal (type II) | AD, AR | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, BMP1 |
| Progressively deforming (type III) | AD, AR | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1, SERPINF1, WNT1 |
| Moderate (type IV) | AD, AR | COL1A1, COL1A2, CRTAP, FKBP10, SP7, SERPINF1, WNT1, TMEM38B |
| With calcification of the interosseous membrane and/or hypertrophic callus (type V) | AD | IFITM5 |
AD, autosomal dominant; AR, autosomal recessive.
In practice, in spite of the complex genotypic variability of OI demonstrated in recent years, its phenotypes are still classified according to Sillence. Genotypic investigation should be indicated, especially in cases suggesting autosomal recessive inheritance, aimed at genetic counseling. The molecular study should be performed using Sanger sequencing of the several new genes, or by next-generation sequencing. Exome sequencing is useful when there is no panel of available genes, or when the involved genes are not known.
FundingCNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico).
Conflicts of interestThe authors declare no conflicts of interest.
To the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for the post-doctoral grant given to Eugênia Ribeiro Valadares in 2013 in the Genetics Sector of the Pediatric Clinic of Freiburg University, Germany, to develop the project “Investigation of osteogenesis imperfecta through the analysis of known genes and new candidate genes in Brazilian and German patients”, under the supervision of Prof. Dr. Bernhard Zabel, Dr. Pablo Villavicencio Lorini, and Dr. Ekkehart Lausch, remarkable individuals.
Please cite this article as: Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics and osteogenesis imperfecta classification? J Pediatr (Rio J). 2014;90:536–41.




