


To characterize cases of suspected congenital disorders of glycosylation (CDG) investigated in a laboratory in southern Brazil using the transferrin isoelectric focusing TfIEF test from 2008 to 2017.
MethodObservational, cross‐sectional, retrospective study. The laboratory records of 1,546 individuals (median age = 36 months, 25‐75 IQR = 10‐108; males = 810) submitted to the TfIEF test during the period were reviewed.
ResultsFifty‐one individuals (3%) had an altered TfIEF pattern (5 ± 2.8 cases/year; median age = 24 months, 25‐75 IQR = 11‐57 months; males = 27, 53%). For 14 of them, data on diagnosis conclusion were available (classic galactosemia = 4; hereditary fructose intolerance = 4; peroxisomal diseases = 2; PMM2‐CDG = 2; MPDU1‐CDG = 1; SLC35A2‐CDG = 1).Comparing the cases with the normal and altered TfIEF patterns, there was a higher prevalence of altered cases in the age group from 11 months to 3 years. There was an increase in the likelihood of change in TfIEF, especially in the presence of inverted nipples or liver disease.
ConclusionsThe data suggest that the investigation of a case with suspected CDG is a complex problem, being aggravated by the existence of other IEMs (inborn errors of metabolism) associated with altered TfIEF pattern and lack of access to confirmatory tests. The presence of inverted nipples and liver disease, especially in individuals aged 11 months to 3 years, should suggest the need for TfIEF investigation.
Caracterizar os casos com suspeita de CDG investigados em laboratório do sul do Brasil pelo exame de IEFTF de 2008 a 2017.
MetodologiaEstudo observacional, transversal, retrospectivo. Foram revisadas as fichas laboratoriais de 1.546 indivíduos (mediana de idade = 36 meses, IQ 25‐75 = 10‐108; sexo masculino = 810) que fizeram o exame de IEFTF no período.
ResultadosCinquenta e um indivíduos (3%) apresentaram padrão alterado na IEFTF (5 ± 2,8 casos/ano; mediana de idade = 24 meses, IQ 25‐75 = 11‐57 meses; sexo masculino = 27, 53%). Para 14 deles, estavam disponíveis dados sobre a conclusão do diagnóstico (galactosemia clássica = 4; intolerância hereditária à frutose = 4; doenças peroxissomais = 2; PMM2‐CDG = 2; MPDU1‐CDG = 1; SLC35A2‐CDG = 1). Comparando os casos com padrão normal e alterado na IEFTF, houve maior prevalência de casos alterados na faixa etária de 11 meses a 3 anos. Verificou‐se um aumento na probabilidade de alteração na IEFTF principalmente na presença de mamilos invertidos ou de hepatopatia.
ConclusõesOs nossos dados sugerem que a investigação de um caso com suspeita de CDG é complexa, é agravada pela existência de outros EIM associados a padrão alterado na IEFTF e pela falta de acesso a exames confirmatórios. A presença principalmente de mamilos invertidos e de hepatopatia em indivíduos na faixa etária de 11 meses a 3 anos deve sugerir a necessidade de investigação por IEFTF.
As doenças congênitas da glicosilação (CDG) são doenças genéticas da síntese e processamentos dos glicanos das glicoproteínas e glicolipídios, de herança autossômica recessiva em sua maioria, caracterizadas pela deficiência total ou parcial de proteínas envolvidas na glicosilação de proteínas ou lipídios.1
Há dois tipos principais de glicosilação de proteínas: N‐glicosilação e O‐ glicosilação. A N‐glicosilação (ligação de N‐glicanos ao grupo amino de asparaginas) compreende uma etapa de montagem e uma etapa de processamento que se estende por três compartimentos: citoplasma, retículo endoplasmático e complexo de Golgi. A O‐glicosilação (ligação de O‐glicanos a grupos hidroxila de treoninas ou serinas) não apresenta etapa de processamento e consiste apenas na etapa de montagem.2 Dessa forma, existem CDG que envolvem N‐glicosilação somente, O‐glicosilação somente ou ambas. Estima‐se que 94% dos indivíduos com CDG apresentem defeitos de N‐glicosilação, os mais frequentes são a PMM2‐CDG (antiga CDG Ia) e a ALG6‐CDG (antiga CDG Ic).3
Desde a primeira descrição clínica em 1980, mais de 100 tipos de CDG foram identificados (tabela 1).3 As manifestações clínicas podem aparecer ainda na gestação, como a hidropisia fetal não imune, microcefalia, malformações congênitas e dismorfias faciais; em neonatos e na infância, podem ser encontrados hipoplasia cerebelar, mamilos invertidos, estrabismo, movimentos oculares anormais, retardo de desenvolvimento neuropsicomotor (RDNPM), hipotonia, ataxia, hiporreflexia, convulsões, anorexia, vômitos, diarreia, defeitos do sistema imunológico, hipogonadismo, lipodistrofia, hepatomegalia, cifose, escoliose e cardiomiopatia. Podem também ocorrer fenótipos mais leves, sem dismorfias e com RNDPM leve. Recomenda‐se, portanto, que a hipótese de CDG seja considerada em qualquer caso de síndrome inexplicada.4
Exemplos de doenças congênitas da glicosilação (CDG) descritas de acordo com sua localização celular e as diferentes vias de glicosilação
| Vias de glicosilação | ||||||
|---|---|---|---|---|---|---|
| Localização celular | N‐Glicosilação | O‐Glicosilação | Lipídica/Glicosilfosfatidilnositol | Múltiplas/OutrasVias | O‐Manosilglicano | |
| Citoplasma | GMPPA‐CDGGMPPB‐CDGMPI‐CDGPMM2‐CDG | EOGT‐CDG | DHDDS‐CDG NANS‐CDGCAD‐CDG PGM1‐CDGGFPT1‐CDG PGM3‐CDGGNE‐CDG CPS2‐CDG | ______ | ||
| Retículo endoplasmático | ALG1‐CDG DPAGT1‐CDGALG2‐CDG MOGS‐CDGALG3‐CDG GANAB‐CDGALG6‐CDG PRKCSH‐CDGALG8‐CDG PRKCSH‐CDGALG9‐CDG RFT1‐CDGALG11‐CDG STT3A‐CDGALG12‐CDG STT3B‐CDGALG13‐CDG SSR3‐CDGALG14‐CDG SSR4‐CDGDDOST‐CDG TUSC3‐CDG | SLC35D1‐CDGPOGLUT1‐CDG | PIGA‐CDGPIGC‐CDGPIGG‐CDGPIGL‐CDGPIGM‐CDGPIGN‐CDGPIGO‐CDG | PIGQ‐CDGPIGT‐CDGPIGV‐CDGPIGW‐CDGPIGY‐CDGPGAP1‐CDG | DOLK‐CDGNUS1‐CDGSRD5A3‐CDGDPM1‐CDGDPM2‐CDGDPM3‐CDGMPDU1‐CDGATP6AP1‐CDGTRAPPC11‐CDG | ______ |
| Golgi | MAN1B1‐CDGMGAT2‐CDG | B4GALT7‐CDG XYLT1‐CDGB3GALT6‐CDG XYLT2‐CDGB3GAT3‐CDG GALNT3‐CDGCHSY1‐CDG LFNG‐CDGEXT1‐CDG POFUT1‐CDGEXT2‐CDGB3GALTL‐CDG | B4GALNT1‐CDGST3GAL5‐CDGPGAP2‐CDGPGAP3‐CDG | B4GALT1‐CDG COG4‐CDGST3GAL3‐CDG COG5‐CDGSLC35A1‐CDG COG6‐CDGSLC35A2‐CDG COG7‐CDGSLC35A3‐CDG COG8‐CDGSLC35C1‐CDG ATP6V0A2‐CDGCOG1‐CDG TIMEM165‐CDGCOG2‐CD VPS138‐CDG | ______ | |
| ERGIC | ______ | ______ | ______ | SEC23B‐CDGCCDC115‐CDGTMEM199‐CDG | ______ | |
| Membrana plasmática | ______ | ______ | ______ | SLC39A8‐CDG | ______ | |
| Membrana do sarcolema | ______ | ______ | ______ | ______ | B3GALNT2‐CDGFKTN‐CDGFKRP‐CDGISPD‐CDGLARGE‐CDGPOMGNT1‐CDGPOMT1‐CDGPOMT2‐CDGTMEM5‐CDGPOMK‐CDG | |
ERGIC, compartimento intermediário do retículo endoplasmático‐Golgi.
Os tipos de CDG em negrito correspondem a casos diagnosticados na presente coorte. A nomenclatura das CDG segue com o nome do gene onde ocorre a mutação hifenizado com CDG.
Fonte dos dados.9–12
Em relação ao diagnóstico, a isoeletrofocalização de transferrina (IEFTF) é um teste de triagem para CDGs que envolvem N‐glicosilação, pois a transferrina é N‐glicosilada. A IEFTF, nesses casos, mostra um deslocamento catódico como consequência da deficiência parcial de ácido siálico.5
A técnica detecta dois padrões de alteração, padrão de alteração do tipo 1, associado principalmente a defeitos relacionados ao retículo endoplasmático (antigamente denominados CDG do tipo I), e o padrão do tipo 2, associado, por sua vez, a defeitos relacionados ao sistema Golgi (antigamente denominados CDG do tipo II).2
No Laboratório de Erros Inatos do Metabolismo do Hospital de Clínicas de Porto Alegre, Rio Grande do Sul (RS), Brasil (LEIM‐HCPA), a IEFTF é o único exame feito para investigação de CDG. O método usado foi adaptado daquele descrito em 1995 por Hackler, Rolf & Kleine.6 Os casos alterados da IEFTF, ou com suspeita de defeitos de O‐glicosilação, devem seguir a investigação para fins de confirmação diagnóstica, seja por ensaios enzimáticos, análises de espectrometria de massa (ESI/MS, MALDI‐TOF) ou análise genética. Frisa‐se que várias condições além de CDG estão associadas a resultados alterados na IEFTF, como galactosemia, intolerância hereditária à frutose,7,8 hepatopatias, uso crônico e abusivo de álcool, infecções por microrganismos produtores de neuraminidase e polimorfismos da transferrina.9–12
No Brasil, pouco se sabe sobre a prevalência ou incidência de CDG na população, bem como sobre a disponibilidade dos exames tanto para a triagem como para o diagnóstico dessa doença. O objetivo deste estudo foi caracterizar os casos triados pelo exame de IEFTF, de 2008 a 2017, no LEIM‐HCPA.
MetodologiaEstudo observacional e retrospectivo de indivíduos investigados por IEFTF no LEIM‐HCPA, de 2008 a 2017. Em 2018, foram coletadas e analisadas informações de fichas laboratoriais dos mesmos. O presente estudo foi aprovado pelo comitê de ética do HCPA com o número 18‐0324.
Coleta de dadosA coleta de dados ocorreu por meio de revisão das fichas do LEIM‐HCPA. Os seguintes dados foram considerados: data da feitura da IEFTF, idade do indivíduo no momento, sexo, local de origem, resultado obtido na IEFTF, sinais e sintomas e informações de histórico familiar. Além disso, para os casos de IEFTF alterados, se houve confirmação do diagnóstico de CDG ou de outra doença mediante exames complementares. Para os indivíduos provenientes do HCPA, também foi feita uma revisão de prontuário eletrônico dos indivíduos com resultados alterados. Foram consideradas como não existentes as informações pertinentes a este estudo que não constavam na ficha laboratorial ou no prontuário eletrônico dos indivíduos.
Análise estatísticaOs bancos de dados foram construídos em Microsoft Office Excel 2010 e para a análise estatística foi usado o programa IBM SPSS Statistics 18.
Os aspectos descritivos foram apresentados nas formas de frequências para os dados correspondentes à IEFTF nos padrões normal e alterado. Médias, desvios‐padrão, medianas e quartis foram usados para os dados de idade. Para a comparação das variáveis entre os grupos com e sem alteração na IEFTF, usou‐se, inicialmente, teste exato de qui‐quadrado de Person, com correção de continuidade, ou exato de Fisher (significância: p < 0,05). Posteriormente, foram feitas análises residuais (razão de prevalência bruta e razão de prevalência ajustada ‐ regressão de Poisson com variância robusta) nos casos com p < 0,05 na análise anterior. Foram consideradas como diferindo entre os grupos com IEFTF alterada ou normal somente as variáveis que apresentaram razão de prevalência ajustada significativa. Para a análise da idade de feitura do teste, foi usado o teste não paramétrico de Mann‐Whitney.
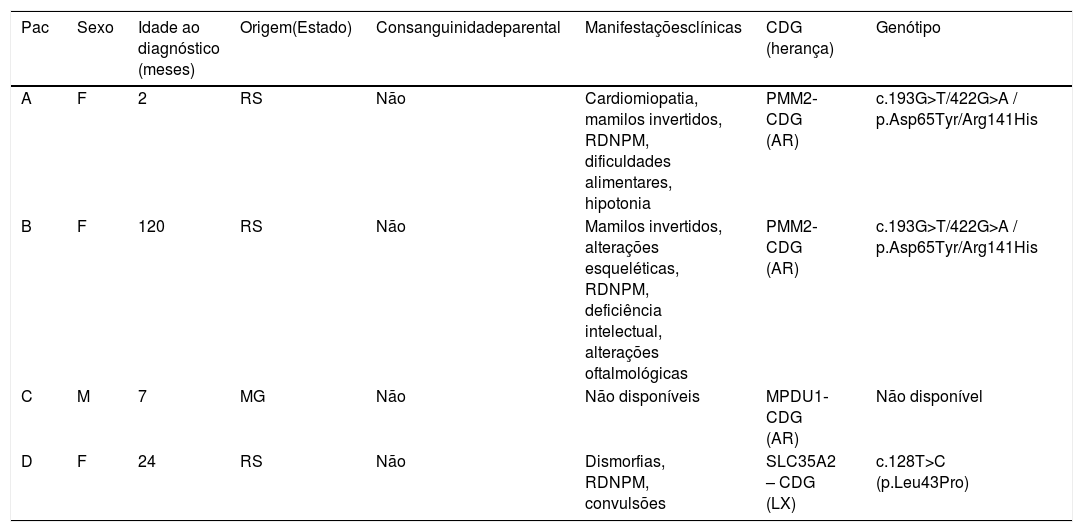
ResultadosDe 2008 a 2017, 1.546 indivíduos fizeram a IEFTF, 566 (37%) provenientes do RS e 980 (63%) de outros estados do Brasil [Santa Catarina/Paraná: 62 (4%); Região Sudeste 744 (48%); Nordeste 77 (5%); Centro‐Oeste 64 (4%); Norte 33 (2%)]. A média anual do número de casos investigados foi de 156 ± 49/casos por ano e, desses, 5 ± 2,8 casos/ano com IEFTF alterada. O maior número de casos investigados foi em 2012 (246) e em 2017 o maior número de casos alterados (11). (fig. 1). Dos indivíduos, 51 (3%; RS = 23) apresentaram alteração no exame IEFTF. Desses, 14 (28%) tiveram a investigação concluída (RS = 11), quatro (RS = 3) chegaram ao diagnóstico confirmatório de CDG por análise genética (MPDU1‐CDG = 1, SLC35A2‐CDG = 1, PMM2‐CDG = 2). Para os casos restantes, exames complementares permitiram a exclusão de CDG e o diagnóstico confirmatório de outras doenças: galactosemia clássica (n = 4, todos confirmados por meio da medida da atividade da Gal1PUT), intolerância hereditária à frutose (n = 4, todos confirmados por análise genética) (tabela 2) e dois casos de doença peroxissomal (ambos confirmados por medida de ácidos graxos de cadeia muito longa). Para os 37 casos restantes, não foi possível a obtenção de informações adicionais acerca da conclusão diagnóstica. Considerando apenas os casos do RS, dos 566 casos investigados por IEFTF, 23 apresentaram padrão alterado (4%) e 11/23 (48%) tiveram o diagnóstico de alguma doença confirmado (CDG = 3/11 ou 27%). A tabela 3 apresenta o perfil completo dos indivíduos incluídos no estudo.
 por ano no Laboratório de Erros Inatos do Metabolismo do Hospital de Clínicas de Porto Alegre, Brasil. Em destaque o número de casos alterados por ano e sua porcentagem.")
Caracterização dos indivíduos com diagnóstico confirmado de doença congênita da glicosilação (n = 4)
| Pac | Sexo | Idade ao diagnóstico (meses) | Origem(Estado) | Consanguinidadeparental | Manifestaçõesclínicas | CDG (herança) | Genótipo |
|---|---|---|---|---|---|---|---|
| A | F | 2 | RS | Não | Cardiomiopatia, mamilos invertidos, RDNPM, dificuldades alimentares, hipotonia | PMM2‐CDG (AR) | c.193G>T/422G>A / p.Asp65Tyr/Arg141His |
| B | F | 120 | RS | Não | Mamilos invertidos, alterações esqueléticas, RDNPM, deficiência intelectual, alterações oftalmológicas | PMM2‐CDG (AR) | c.193G>T/422G>A / p.Asp65Tyr/Arg141His |
| C | M | 7 | MG | Não | Não disponíveis | MPDU1‐CDG (AR) | Não disponível |
| D | F | 24 | RS | Não | Dismorfias, RDNPM, convulsões | SLC35A2 – CDG (LX) | c.128T>C (p.Leu43Pro) |
AR, autossômica recessiva; F, feminino; LX, ligada ao X; M, masculino; MG, Minas Gerais; Pac, paciente; RDNPM, retardo do desenvolvimento neuropsicomotor; RS, Rio Grande do Sul.
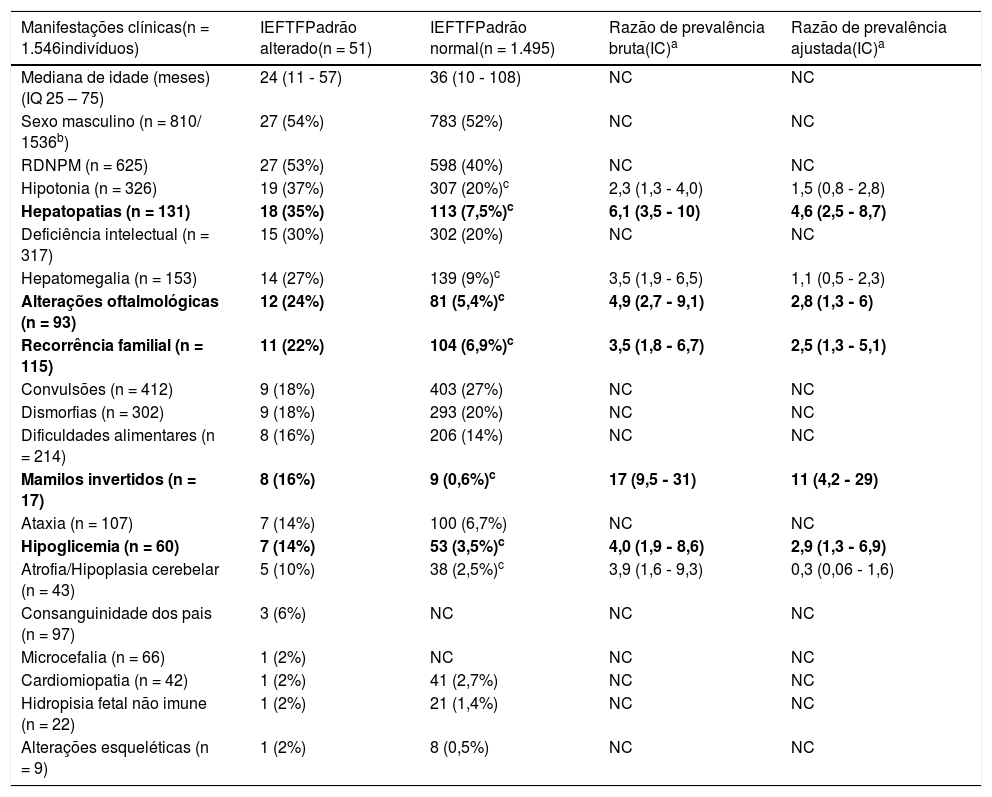
Resumo dos casos avaliados por isoeletrofocalização da transferrina em soro no Laboratório de Erros Inatos do Metabolismo do Hospital de Clínicas de Porto Alegre, Brasil (2008‐2017)
| Manifestações clínicas(n = 1.546indivíduos) | IEFTFPadrão alterado(n = 51) | IEFTFPadrão normal(n = 1.495) | Razão de prevalência bruta(IC)a | Razão de prevalência ajustada(IC)a |
|---|---|---|---|---|
| Mediana de idade (meses) (IQ 25 – 75) | 24 (11 ‐ 57) | 36 (10 ‐ 108) | NC | NC |
| Sexo masculino (n = 810/ 1536b) | 27 (54%) | 783 (52%) | NC | NC |
| RDNPM (n = 625) | 27 (53%) | 598 (40%) | NC | NC |
| Hipotonia (n = 326) | 19 (37%) | 307 (20%)c | 2,3 (1,3 ‐ 4,0) | 1,5 (0,8 ‐ 2,8) |
| Hepatopatias (n = 131) | 18 (35%) | 113 (7,5%)c | 6,1 (3,5 ‐ 10) | 4,6 (2,5 ‐ 8,7) |
| Deficiência intelectual (n = 317) | 15 (30%) | 302 (20%) | NC | NC |
| Hepatomegalia (n = 153) | 14 (27%) | 139 (9%)c | 3,5 (1,9 ‐ 6,5) | 1,1 (0,5 ‐ 2,3) |
| Alterações oftalmológicas (n = 93) | 12 (24%) | 81 (5,4%)c | 4,9 (2,7 ‐ 9,1) | 2,8 (1,3 ‐ 6) |
| Recorrência familial (n = 115) | 11 (22%) | 104 (6,9%)c | 3,5 (1,8 ‐ 6,7) | 2,5 (1,3 ‐ 5,1) |
| Convulsões (n = 412) | 9 (18%) | 403 (27%) | NC | NC |
| Dismorfias (n = 302) | 9 (18%) | 293 (20%) | NC | NC |
| Dificuldades alimentares (n = 214) | 8 (16%) | 206 (14%) | NC | NC |
| Mamilos invertidos (n = 17) | 8 (16%) | 9 (0,6%)c | 17 (9,5 ‐ 31) | 11 (4,2 ‐ 29) |
| Ataxia (n = 107) | 7 (14%) | 100 (6,7%) | NC | NC |
| Hipoglicemia (n = 60) | 7 (14%) | 53 (3,5%)c | 4,0 (1,9 ‐ 8,6) | 2,9 (1,3 ‐ 6,9) |
| Atrofia/Hipoplasia cerebelar (n = 43) | 5 (10%) | 38 (2,5%)c | 3,9 (1,6 ‐ 9,3) | 0,3 (0,06 ‐ 1,6) |
| Consanguinidade dos pais (n = 97) | 3 (6%) | NC | NC | NC |
| Microcefalia (n = 66) | 1 (2%) | NC | NC | NC |
| Cardiomiopatia (n = 42) | 1 (2%) | 41 (2,7%) | NC | NC |
| Hidropisia fetal não imune (n = 22) | 1 (2%) | 21 (1,4%) | NC | NC |
| Alterações esqueléticas (n = 9) | 1 (2%) | 8 (0,5%) | NC | NC |
IC, intervalo de confiança; IETFTF, isoeletrofocalização da transferrina; M, sexo masculino; NC, não calculado; RDNPM, retardo do desenvolvimento neuropsicomotor.
a Comparação feita entre os casos com padrão normal e padrão alterado na IEFTF.
b 10 casos que não tiveram o sexo do indivíduo informado.
Para as comparações, inicialmente foi usado o teste do qui‐quadrado de Person com correção de continuidade, ou o teste exato de Fisher (c resultados significativos, p < 0,05); após, foi calculada a razão de prevalência bruta para as variáveis com p < 0,05 e, para as variáveis com diferenças significativas na razão de prevalência bruta, foi calculada a razão de prevalência ajustada. As variáveis em negrito são aquelas que apresentaram razão de prevalência ajusta estatisticamente significativa.
A faixa de 11 a 36 meses foi a que apresentou a maior prevalência de resultados alterados, 48% dos indivíduos que apresentaram resultado alterado encontram‐se nessa faixa etária.
A manifestação clínica mais comum nos casos investigados foi RDNPM, seguido de convulsões, hipotonia, deficiência intelectual e dismorfias. Porém, os sintomas mais frequentes nos casos com alterações na IEFTF foram: RDNPM, hipotonia, hepatopatia, deficiência intelectual e hepatomegalia, foram mamilos invertidos (razão de prevalência ajustada = 11), seguidos de hepatopatia, hipoglicemia, alterações oftalmológicas e recorrência familial às manifestações clínicas estatisticamente associadas à IEFTF alterada (tabela 3).
DiscussãoCDGs têm sido descritas como um grande desafio diagnóstico, uma vez que, nesse conjunto de doenças, quase todos os órgãos são acometidos e uma grande variedade de sintomas já foi descrita. Assim, a recomendação é considerar CDG em qualquer caso de doença não explicada, principalmente quando há presença de sintomas neurológicos.13 No Brasil, um outro fator que contribui para a dificuldade diagnóstica é a dificuldade de acesso a exames confirmatórios. Dentro desse contexto, nossos dados sugerem que existe subdiagnóstico de CDG no Brasil e que esforços devem ser feitos para a implantação dos testes diagnósticos.
O protocolo de investigação para CDG estabelecido no HCPA não contempla uma investigação completa para a doença, uma vez que somente a IEFTF é oferecida. O protocolo mais custo‐efetivo, após terem sido descartadas outras doenças que possam causar alteração da IEFTF, não está bem estabelecido. Uma estratégia possível seria o sequenciamento inicial apenas do gene PMM2 para os casos alterados com padrão de CDG tipo I, uma vez que esses são os casos mais frequentes de CDG. Se o sequenciamento não evidenciar anormalidade, uma análise de painel com genes específicos para CDG e, em caso de resultado negativo, o sequenciamento completo do exoma estariam indicados.3 Outra estratégia possível seria a investigação inicial direta por meio de painel incluídos os genes causadores de CDG.
Apesar de as CDG serem doenças genéticas raras, existe um número significativo de indivíduos com suspeita da doença, como pode ser visto pela média anual de casos investigados por IEFTF no LEIM‐HCPA. Entretanto, o número de casos confirmados foi pequeno (apenas 4 indivíduos em dez anos), o que sugere a ocorrência de um subdiagnóstico dessas doenças em nosso país, ainda mais considerando que a frequência estimada para PMM2‐CDG, que vem ser a CDG mais comum, é de cerca de 1/20.000 recém‐nascidos.14 Mesmo na ausência de tratamento específico para a maioria das CDG, a confirmação do seu diagnóstico é fundamental para o aconselhamento genético, haja vista o elevado risco de recorrência (a maioria das CDG apresenta padrão de herança autossômico recessivo ou ligado ao X).
Nosso estudo mostrou, também, que galactosemia clássica e intolerância hereditária à frutose são causas frequentes de alterações na IEFTF. Galactosemia tem sido referida como um segundo tipo de defeito de glicosilação.5,7 A literatura aponta que há anormalidades na glicosilação de N‐glicanos na transferrina sérica em neonatos com galactosemia não tratados. Essas anormalidades, em sua grande parte, são resolvidas com o tratamento dietético com restrição de galactose.7,15,16 Outra doença que pode acarretar em hipoglicosilação das isoformas de transferrina é a intolerância hereditária à frutose. É causada por uma deficiência na atividade da enzima aldolase B; consequentemente, a frutose‐1P é acumulada em células hepáticas, acarreta a inibição da atividade da fosfomanose isomerase, enzima presente nas primeiras etapas da glicosilação de proteínas.17,18
Por outro lado, tendo‐se em consideração que o fígado é um dos principais órgãos de glicosilação, também responsável pela produção da maioria das proteínas séricas glicosiladas, defeitos de glicosilação estão associados a sintomas clínicos hepáticos, os quais estão presentes em torno de 20% dos casos de CDG.19 Nosso estudo corrobora esses achados, visto que as hepatopatias apresentam forte associação com alterações no padrão da IEFTF (indivíduos com hepatopatia têm 4,6 vezes mais chances de apresentar uma alteração na IEFTF do que indivíduos sem esse sintoma). Hepatopatias estão associadas a resultados alterados na IEFTF, porém essa alteração pode não estar diretamente associada às CDG, e sim a outros EIM que afetam o fígado, como as doenças peroxissomais, identificadas em dois casos com alteração da IEFTF no nosso estudo.
O fato de que faixa de 11 a 36 meses foi a que apresentou a maior prevalência de resultados alterados da IEFTF sugere que as condições causadoras de alterações na IEFTF (entre elas, as CDG) apresentam‐se principalmente na infância, após o período de lactância.
Nossos dados mostram que a presença de mamilos invertidos apresentou o maior índice de associação com a alteração da IEFTF (a razão de prevalência ajustada demonstra que esse sintoma aumenta em 11 vezes a chance de o indivíduo apresentar a IEFTF alterada). Mamilos invertidos estão geralmente presentes em indivíduos com CDG ao nascimento, podem voltar à posição normal em poucos dias ou semanas de vida. Entretanto, a persistência do achado na vida adulta pode ocorrer. É importante salientar, no entanto, que a lista de síndromes associadas a mamilos invertidos é extensa, inclui, por exemplo, síndromes de Turner, de Smith‐Lemli‐Optiz e de Robinow, entre outras, porém, também pode ser observado em indivíduos normais. No caso das CDG, os mamilos invertidos são encontrados com maior frequência em indivíduos com defeitos de N‐glicosilação.20
Com relação às limitações do estudo, é importante mencionar que está baseado em informações clínicas presentes nos prontuários dos indivíduos com suspeita de CDG. Outra limitação importante, com relação aos indivíduos provenientes de outras regiões do país, é que, após a liberação do laudo aos serviços de origem do paciente, não temos informações do seguimento que esses indivíduos tiveram, ou seja, se o paciente concluiu a investigação ou não.
ConclusãoApesar das limitações do estudo no que diz respeito à análise retrospectiva dos casos, mediante revisão de prontuários e a falta de uniformização do atendimento e diagnóstico dos pacientes provenientes de diferentes regiões do país, verificou‐se que há necessidade da implantação de novas técnicas para diagnóstico de CDG e o estabelecimento de um protocolo mais completo para a investigação das CDG no LEIM‐HCPA. Os achados mostram que, apesar de alterações na IEFTF terem sido reportadas em 51 casos, conseguimos constatar em apenas quatro indivíduos o diagnóstico confirmado para CDG.
Os dados obtidos também mostram que os principais sintomas que levaram à suspeita de CDG foram neurológicos. No entanto, os indivíduos que apresentarem mamilos invertidos, hepatopatia, hipoglicemia, alterações oftalmológicas e a presença de outros casos na família foram os que tiveram maior probabilidade de ter IEFTF alterada. Dessa forma, a triagem para CDG deve ser especialmente considerada quando houver a presença dessas manifestações clínicas.
FinanciamentoFundo de Incentivo à Pesquisa e Eventos do Hospital de Clínicas de Porto Alegre (FIPE‐HCPA), número do financiamento: 18‐0324.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Aos colegas Zeniara Lompa, Régis Guidobono, Karen Lucas e Tânia Braga, funcionários do Serviço de Genética Médica do HCPA, pelo apoio no estudo. Somos gratos também ao apoio financeiro fornecido pelo FIPE‐HCPA.
Como citar este artigo: Magalhães AP, Burin MG, Souza CF, Bitencourt FH, Sebastião FM, Silva TO, et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten‐year experience in a Brazilian center. J Pediatr (Rio J). 2020;96:710–6.
Estudo feito na Universidade Federal do Rio Grande do Sul (UFRGS), Hospital de Clínicas de Porto Alegre (HCPA), Porto Alegre, RS, Brasil.




