


To characterize cases of suspected congenital disorders of glycosylation (CDG) investigated in a laboratory in southern Brazil using the transferrin isoelectric focusing TfIEF test from 2008 to 2017.
MethodObservational, cross-sectional, retrospective study. The laboratory records of 1,546 individuals (median age=36 months, 25–75 IQR=10–108; males=810) submitted to the TfIEF test during the period were reviewed.
ResultsFifty-one individuals (3%) had an altered TfIEF pattern (5±2.8 cases/year; median age=24 months, 25–75 IQR=11–57 months; males=27, 53%). For 14 of them, data on diagnosis conclusion were available (classic galactosemia=4; hereditary fructose intolerance=4; peroxisomal diseases=2; PMM2-CDG=2; MPDU1-CDG=1; SLC35A2-CDG=1).Comparing the cases with the normal and altered TfIEF patterns, there was a higher prevalence of altered cases in the age group from 11 months to 3 years. There was an increase in the likelihood of change in TfIEF, especially in the presence of inverted nipples or liver disease.
ConclusionsThe data suggest that the investigation of a case with suspected CDG is a complex problem, being aggravated by the existence of other IEMs (inborn errors of metabolism) associated with altered TfIEF pattern and lack of access to confirmatory tests. The presence of inverted nipples and liver disease, especially in individuals aged 11 months to 3 years, should suggest the need for TfIEF investigation.
Caracterizar os casos com suspeita de CDG investigados em laboratório do sul do Brasil pelo exame de IEFTF de 2008 a 2017.
MetodologiaEstudo observacional, transversal, retrospectivo. Foram revisadas as fichas laboratoriais de 1.546 indivíduos (mediana de idade=36 meses, IQ 25-75 =10-108; sexo masculino=810) que fizeram o exame de IEFTF no período.
ResultadosCinquenta e um indivíduos (3%) apresentaram padrão alterado na IEFTF (5±2,8 casos/ano; mediana de idade=24 meses, IQ 25-75 =11-57 meses; sexo masculino=27, 53%). Para 14 deles, estavam disponíveis dados sobre a conclusão do diagnóstico (galactosemia clássica=4; intolerância hereditária à frutose=4; doenças peroxissomais=2; PMM2-CDG=2; MPDU1-CDG=1; SLC35A2-CDG=1). Comparando os casos com padrão normal e alterado na IEFTF, houve maior prevalência de casos alterados na faixa etária de 11 meses a 3 anos. Verificou-se um aumento na probabilidade de alteração na IEFTF principalmente na presença de mamilos invertidos ou de hepatopatia.
ConclusõesOs nossos dados sugerem que a investigação de um caso com suspeita de CDG é complexa, é agravada pela existência de outros EIM associados a padrão alterado na IEFTF e pela falta de acesso a exames confirmatórios. A presença principalmente de mamilos invertidos e de hepatopatia em indivíduos na faixa etária de 11 meses a 3 anos deve sugerir a necessidade de investigação por IEFTF.
Congenital disorders of glycosylation (CDG) are genetic diseases that affect the synthesis and processing of glycans from glycoproteins and glycolipids, of autosomal recessive inheritance for the most part, characterized by total or partial deficiency of proteins involved in protein or lipid glycosylation.1
There are two main types of protein glycosylation: N-glycosylation and O-glycosylation. N-glycosylation (binding of N-glycans to the amino group of asparagine) comprises an assembly step and a processing step that covers three distinct compartments: cytoplasm, endoplasmic reticulum, and Golgi complex. O-glycosylation (binding of O-glycans to threonine or serine hydroxyl groups) has no processing step and consists only of the assembly step.2 Thus, there are CDGs that involve N-glycosylation only, O-glycosylation only, or both. It is estimated that 94% of individuals with CDG have N-glycosylation defects, with the most frequent being PMM2-CDG (formerly CDG Ia) and ALG6-CDG (formerly CDG Ic).3
Since the first clinical description in 1980, more than 100 types of CDGs have been identified (Table 1).3 Clinical manifestations can appear as early as intrauterine life, such as nonimmune fetal hydrops, microcephaly, congenital malformations, and facial dysmorphisms; in neonates and childhood, cerebellar hypoplasia, inverted nipples, strabismus, abnormal eye movements, neuropsychomotor developmental delay (NPMD), hypotonia, ataxia, hyporeflexia, seizures, anorexia, vomiting, diarrhea, immune system defects, hypogonadism, lipodystrophy, hepatomegaly, kyphosis, scoliosis, and cardiomyopathy can occur. Milder non-dysmorphic features may also occur. It is therefore recommended that the hypothesis of CDG be considered in any case of unexplained syndrome.4
Examples of congenital disorders of glycosylation (CDG) described according to their cell location and the different glycosylation pathways.
| GLYCOSYLATION PATHWAYS | |||||||
|---|---|---|---|---|---|---|---|
| CELL LOCATION | N-glycosylation | O-glycosylation | Lipidic/glycosylphosphatidylinositol | Multiples/other pathways | O-mannosylglycan | ||
| CYTOPLASM | GMPPA-CDGGMPPB-CDGMPI-CDGPMM2-CDG | EOGT-CDG | DHDDS-CDG NANS-CDGCAD-CDG PGM1-CDGGFPT1-CDG PGM3-CDGGNE-CDG CPS2-CDG | — | |||
| ENDOPLASMIC RETICULUM | ALG1-CDG DPAGT1-CDGALG2-CDG MOGS-CDGALG3-CDG GANAB-CDGALG6-CDG PRKCSH-CDGALG8-CDG PRKCSH-CDGALG9-CDG RFT1-CDGALG11-CDG STT3A-CDGALG12-CDG STT3B-CDGALG13-CDG SSR3-CDGALG14-CDG SSR4-CDGDDOST-CDG TUSC3-CDG | SLC35D1-CDGPOGLUT1-CDG | PIGA-CDGPIGC-CDGPIGG-CDGPIGL-CDGPIGM-CDGPIGN-CDGPIGO-CDG | PIGQ-CDGPIGT-CDGPIGV-CDGPIGY-CDGPGAP1-CDG | DOLK-CDGNUS1-CDGSRD5A3-CDGDPM1-CDGDPM2-CDGDPM3-CDGMPDU1-CDGATP6AP1-CDGTRAPPC11-CDG | — | |
| GOLGI APPARATUS | MAN1B1-CDGMGAT2-CDG | B4GALT7-CDGB3GALT6-CDGB3GAT3-CDGCHSY1-CDGEXT1-CDGEXT2-CDGB3GALTL-CDG | XYLT1-CDGXYLT2-CDGGALNT3-CDGLFNG-CDGPOFUT1-CDG | B4GALNT1-CDGST3GAL5-CDGPGAP2-CDGPGAP3-CDG | B4GALT1-CDG COG4-CDGST3GAL3-CDG COG5-CDGSLC35A1-CDG COG6-CDGSLC35A2-CDGCOG7-CDGSLC35A3-CDG COG8-CDGSLC35C1-CDG ATP6V0A2-CDGCOG1-CDG TIMEM165-CDGCOG2-CD VPS138-CDG | — | |
| ERGIC | — | — | — | SEC23B-CDGCCDC115-CDGTMEM199-CDG | — | ||
| PLASMA MEMBRANE | — | — | — | SLC39A8-CDG | — | ||
| SARCOLEMA MEMBRANE | — | — | — | — | B3GALNT2-CDGFKTN-CDGFKRP-CDGISPD-CDGLARGE-CDGPOMGNT1-CDGPOMT1-CDGPOMT2-CDGTMEM5-CDGPOMK-CDG | ||
ERGIC, endoplasmic reticulum golgi intermediate compartment.
The CDG types in bold correspond to cases diagnosed in the present cohort. The CDG nomenclature follows the name of the gene where the mutation occurs, hyphenated with CDG.
Source of data.9–12
Regarding the diagnosis, transferrin isoelectric focusing (TfIEF) is a screening test for CDGs that involves N-glycosylation, because transferrin is N-glycosylated. The TfIEF in these cases shows a cathodic displacement as a consequence of partial sialic acid deficiency.5
The technique detects two alteration patterns; the type 1 alteration pattern, mainly associated with defects related to the endoplasmic reticulum (formerly known as type I CDG), and the type 2 pattern, in turn, associated with defects related to the Golgi system (formerly called type II CDG).2
At the Laboratory of Inborn Errors of Metabolism of Hospital de Clínicas de Porto Alegre, Rio Grande do Sul (RS), Brazil (LEIM-HCPA), the TfIEF is the only test performed for CDG investigation. The method used was adapted from that described in 1995 by Hackler, Rolf & Kleine.6 Cases with altered TfIEF, or suspected of O-glycosylation defects, should be submitted to investigation for diagnostic confirmation, either by enzymatic testing, electrospray ionization—mass spectrometry/matrix-assisted laser desorption/ionization—time-of-flight mass spectrometry,(ESI/MS, MALDI-TOF), or genetic analysis. It is noteworthy that several conditions besides CDG are associated with altered TfIEF results, such as galactosemia, hereditary fructose intolerance,7,8 liver disease, chronic alcohol abuse, infections by neuraminidase-producing microorganisms, and transferrin polymorphisms.9–12
In Brazil, little is known about the prevalence or incidence of CDG in the population, nor about the availability of tests for both screening and diagnosis of this disease. The aim of this study is to characterize the cases screened by TfIEF at the LEIM-HCPA, from 2008 to 2017.
MethodsThis was an observational and retrospective study of individuals investigated by TfIEF at LEIM-HCPA, from 2008 to 2017. In 2018, information from these cases was collected from their laboratory files and analyzed. This study was approved by the HCPA Ethics Committee under number 18-0324.
Data collectionData collection was carried out by reviewing LEIM-HCPA files. The following data were considered: date of TfIEF performance, individual’s age, gender, place of origin, TfIEF result, signs and symptoms, and family history. Moreover, for altered TfIEF cases, the diagnosis of CDG or another disease was confirmed by complementary tests. For individuals coming from the HCPA, an electronic medical record review was also performed for individuals with altered results. Information relevant to this study that was not found in the laboratory file or in the individuals' electronic medical record was considered non-existent.
Statistical analysisThe databases were created using Microsoft Office Excel (Microsoft Corporation, Excel, version 2010, WA, USA) and the IBM SPSS Statistics (Released 2009. PASW Statistics for Windows, version 18.0, IL, USA) program was used for the statistical analysis.
The descriptive aspects were presented as frequencies for data corresponding to the normal and altered patterns of TfIEF. Data on age was presented as means, standard deviations, medians, and quartiles. Pearson’s exact chi-squared test was initially used to compare the variables between the groups with and without TfIEF alterations, with continuity correction, or Fisher's exact test (significance: p<0.05). Subsequently, residual analyses (crude prevalence ratio and adjusted prevalence ratio—Poisson regression with robust variance) were performed in cases with p<0.05 in the previous analysis. Only variables with a significant adjusted prevalence ratio were considered as differing between the groups with altered or normal TfIEF. For the analysis of the age when the test was performed, the nonparametric Mann-Whitney test was used.
ResultsFrom 2008 to 2017, 1546 individuals underwent TfIEF, of whom 566 (37%) were from RS and 980 (63%) from other states in Brazil (Santa Catarina/Paraná: 62 [4%]; Southeast region 744 [48%]; Northeast 77 [5%]; Midwest 64 [4%]; North 33 [2%]). The annual average number of investigated cases was 156±49 cases per year, and of these, 5±2.8 cases per year with altered TfIEF. The highest number of investigated cases was observed in 2012 (246); in 2017, the highest number of altered cases (11) was observed. (Fig. 1) Fifty-one individuals (3%; RS=23) showed alterations in the TfIEF test. Of these, 14 (28%) completed the investigation (RS=11), and four individuals (RS=3) attained the confirmatory diagnosis of CDG through genetic analysis (MPDU1-CDG=1, SLC35A2-CDG=1, PMM2-CDG=2). For the remaining cases, complementary tests allowed ruling out CDG and the confirmatory diagnosis of other diseases: classic galactosemia (n=4, all confirmed by measuring Galactose-1-phosphate Uridyl Transferase activity (Gal1PUT)), hereditary fructose intolerance (n=4, all confirmed by genetic analysis) (Table 2), and two cases of peroxisomal disease (both confirmed by measurement of very long-chain fatty acids).For the remaining 37 cases, it was not possible to obtain additional information about the diagnostic conclusion. Considering only the cases from RS, of the 566 cases investigated by TfIEF, 23 had an altered pattern (4%) and 11/23 (48%) had a confirmed disease diagnosis (CDG=3/11 or 27%). Table 3 shows the complete profile of the individuals included in the study.
 per year at the Metabolism Inborn Errors Laboratory of the Hospital de Clínicas de Porto Alegre, Brazil. The number of altered cases per year and their percentage are highlighted.")
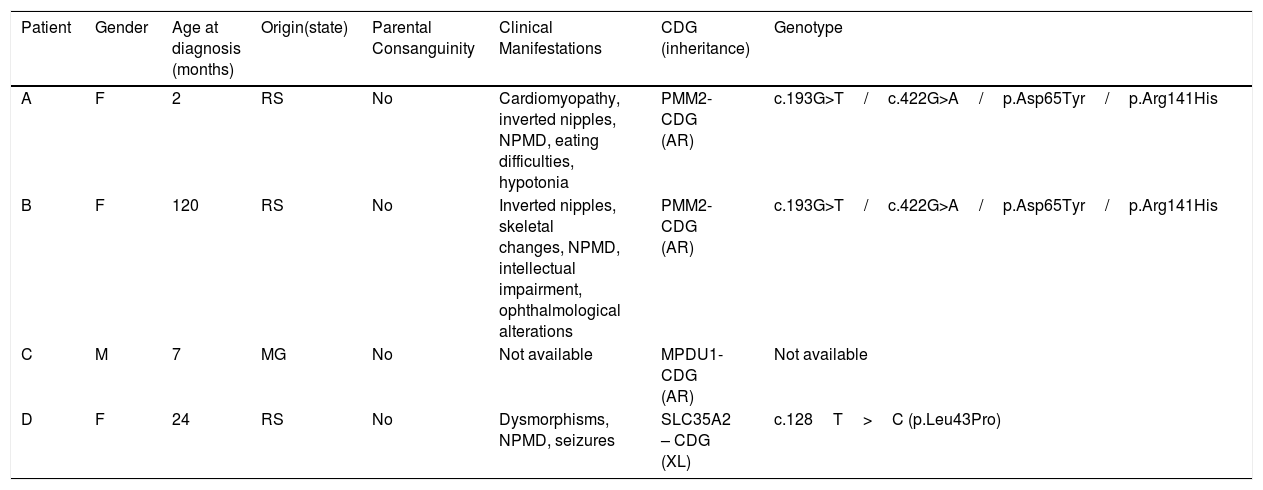
Characterization of individuals with a confirmed diagnosis of congenital disorders of glycosylation (n=4).
| Patient | Gender | Age at diagnosis (months) | Origin(state) | Parental Consanguinity | Clinical Manifestations | CDG (inheritance) | Genotype |
|---|---|---|---|---|---|---|---|
| A | F | 2 | RS | No | Cardiomyopathy, inverted nipples, NPMD, eating difficulties, hypotonia | PMM2-CDG (AR) | c.193G>T/c.422G>A/p.Asp65Tyr/p.Arg141His |
| B | F | 120 | RS | No | Inverted nipples, skeletal changes, NPMD, intellectual impairment, ophthalmological alterations | PMM2-CDG (AR) | c.193G>T/c.422G>A/p.Asp65Tyr/p.Arg141His |
| C | M | 7 | MG | No | Not available | MPDU1-CDG (AR) | Not available |
| D | F | 24 | RS | No | Dysmorphisms, NPMD, seizures | SLC35A2 – CDG (XL) | c.128T>C (p.Leu43Pro) |
Patients A and B are not related. F, female; M, male; NPMD, neuro psycho motor developmental delay; RS, Rio Grande do Sul; MG, Minas Gerais; AR, autosomal recessive; XL, X-linked.
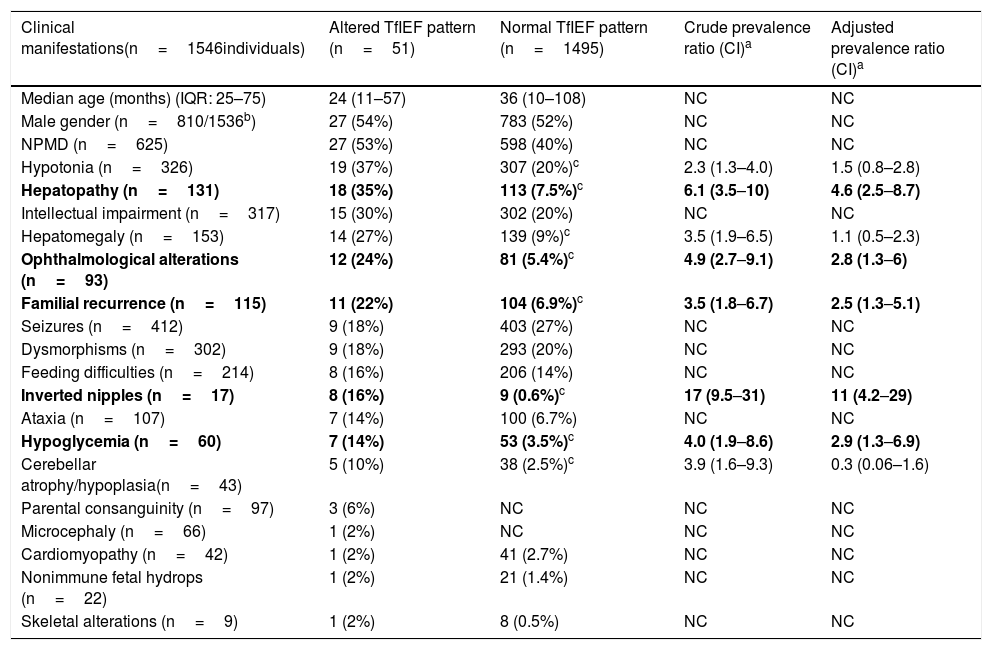
Summary of cases evaluated by serum transferrin isoelectric focusing at the Laboratory of Inborn Errors of Metabolism of Hospital de Clínicas de Porto Alegre, Brazil (2008–2017).
| Clinical manifestations(n=1546individuals) | Altered TfIEF pattern (n=51) | Normal TfIEF pattern (n=1495) | Crude prevalence ratio (CI)a | Adjusted prevalence ratio (CI)a |
|---|---|---|---|---|
| Median age (months) (IQR: 25–75) | 24 (11–57) | 36 (10–108) | NC | NC |
| Male gender (n=810/1536b) | 27 (54%) | 783 (52%) | NC | NC |
| NPMD (n=625) | 27 (53%) | 598 (40%) | NC | NC |
| Hypotonia (n=326) | 19 (37%) | 307 (20%)c | 2.3 (1.3–4.0) | 1.5 (0.8–2.8) |
| Hepatopathy (n=131) | 18 (35%) | 113 (7.5%)c | 6.1 (3.5–10) | 4.6 (2.5–8.7) |
| Intellectual impairment (n=317) | 15 (30%) | 302 (20%) | NC | NC |
| Hepatomegaly (n=153) | 14 (27%) | 139 (9%)c | 3.5 (1.9–6.5) | 1.1 (0.5–2.3) |
| Ophthalmological alterations (n=93) | 12 (24%) | 81 (5.4%)c | 4.9 (2.7–9.1) | 2.8 (1.3–6) |
| Familial recurrence (n=115) | 11 (22%) | 104 (6.9%)c | 3.5 (1.8–6.7) | 2.5 (1.3–5.1) |
| Seizures (n=412) | 9 (18%) | 403 (27%) | NC | NC |
| Dysmorphisms (n=302) | 9 (18%) | 293 (20%) | NC | NC |
| Feeding difficulties (n=214) | 8 (16%) | 206 (14%) | NC | NC |
| Inverted nipples (n=17) | 8 (16%) | 9 (0.6%)c | 17 (9.5–31) | 11 (4.2–29) |
| Ataxia (n=107) | 7 (14%) | 100 (6.7%) | NC | NC |
| Hypoglycemia (n=60) | 7 (14%) | 53 (3.5%)c | 4.0 (1.9–8.6) | 2.9 (1.3–6.9) |
| Cerebellar atrophy/hypoplasia(n=43) | 5 (10%) | 38 (2.5%)c | 3.9 (1.6–9.3) | 0.3 (0.06–1.6) |
| Parental consanguinity (n=97) | 3 (6%) | NC | NC | NC |
| Microcephaly (n=66) | 1 (2%) | NC | NC | NC |
| Cardiomyopathy (n=42) | 1 (2%) | 41 (2.7%) | NC | NC |
| Nonimmune fetal hydrops (n=22) | 1 (2%) | 21 (1.4%) | NC | NC |
| Skeletal alterations (n=9) | 1 (2%) | 8 (0.5%) | NC | NC |
TfIEF, transferrin isoelectric focusing; M, male gender; NPMD, neuropsychomotor developmental delay; CI, confidence interval; NC, not calculated.
For comparisons, Pearson's chi-squared test was initially used with continuity correction, or Fisher's exact test (csignificant results, p<0.05); afterwards, the crude prevalence ratio was calculated for the variables with p<0.05 and for the variables with significant differences in the crude prevalence ratio, the adjusted prevalence ratio was calculated. The variables in bold are those with a statistically significant adjusted prevalence ratio.
aComparison between cases with normal pattern and altered pattern in TfIEF.
The age group from 11 to 36 months was the group with the highest prevalence of altered results, as 48% of individuals with altered results were in this age group.
The most common clinical manifestation in the investigated cases was NPMD, followed by seizures, hypotonia, intellectual impairment, and dysmorphisms. However, the most frequent symptoms in cases with alterations in the TfIEF were: NPMD, hypotonia, liver disease, intellectual impairment, and hepatomegaly, with inverted nipples (adjusted prevalence ratio=11), followed by liver disease, hypoglycemia, ophthalmic alterations, and familial recurrence representing the clinical manifestations statistically associated with altered TfIEF (Table 3).
DiscussionCDGs have been described as a major diagnostic challenge, since in this set of diseases almost all organs are affected, and a wide variety of symptoms have already been described. Therefore, the recommendation is to consider CDG in any case of unexplained disease, especially in the presence of neurological symptoms.13 In Brazil, another factor that contributes to diagnostic difficulty is the problems in accessing confirmatory tests. Within this context, these data suggest the underdiagnosis of CDGs in Brazil, and that efforts should be made to implement the diagnostic tests.
The protocol for CDG investigation established in HCPA does not include a full investigation of the disease, as only TfIEF is offered. The most cost-effective protocol, after ruling out other diseases that may cause TfIEF alteration, is not well established yet. A possible strategy would be the initial sequencing of the PMM2 gene only for altered cases with the type I CDG pattern, since these are the most frequent cases of CDG. If the sequencing does not disclose any abnormality, a panel analysis with CDG-specific genes would be performed and, if negative, a full exome sequencing would be indicated.3 Another possible strategy would be a direct initial investigation through a panel including the genes that cause CDG.
Although CDGs are rare genetic diseases, there is a significant number of individuals with suspected disease, as shown by the annual average of cases investigated by TfIEF in LEIM-HCPA. However, the number of confirmed cases was small (only four individuals in ten years), suggesting the occurrence of the underdiagnosis of these diseases in Brazil, especially considering that the estimated frequency for PMM2-CDG, which is the most common CDG, is approximately 1:20,000 newborns.14 Even in the absence of specific treatment for most CDGs, diagnostic confirmation is critical for genetic counseling, given the high risk of recurrence (most CDGs have an X-linked or recessive autosomal inheritance pattern).
This study also showed that classical galactosemia and hereditary fructose intolerance are frequent causes of TfIEF alterations. Galactosemia has been reported as a second type of glycosylation defect.5,7 The literature indicates there are abnormalities in the glycosylation of N-glycans in the serum transferrin of neonates with untreated galactosemia. These abnormalities are largely resolved by dietary treatment with galactose restriction.7,15,16 Another disease that can lead to the hypoglycosylation of transferrin isoforms is hereditary fructose intolerance. It is caused by a deficiency in aldolase B enzyme activity and, therefore, fructose-1P accumulates in liver cells, resulting in the inhibition of phosphomannose isomerase, an enzyme present in the early stages of protein glycosylation.17,18
However, considering the liver is the major glycosylation organ, also responsible for the production of most glycosylated serum proteins, glycosylation defects are associated with clinical liver symptoms, which are present in approximately 20% of CDG cases.19 The present study corroborates these findings, as liver disease is strongly associated with changes in the TfIEF pattern (individuals with liver disease are 4.6-fold more likely to have an alteration in TfIEF than individuals without this symptom). Liver diseases are associated with altered TfIEF results; however, this alteration may not be directly associated with CDGs, but rather with other IEMs that affect the liver, such as peroxisomal diseases, identified in two cases with altered TfIEF in our study.
The fact that the age group of 11–36 months showed the highest prevalence of altered TfIEF results suggests that the conditions that cause TfIEF alterations (among them the CDGs) occur mainly in childhood after the breastfeeding period.
These data show that the presence of inverted nipples had the highest rate of association with TfIEF alteration (the adjusted prevalence ratio shows that this symptom increases the individual's chance of having altered TfIEF by 11-fold). Inverted nipples are usually present in individuals with CDG at birth and may return to normal within a few days or weeks of life. However, persistence of the finding in adulthood may occur. It is important to note, however, that the list of syndromes associated with inverted nipple is a long one, including, for instance, Turner, Smith-Lemli-Optiz, and Robinow syndromes, among others; nonetheless, it can also be observed in normal individuals. In the case of CDG, inverted nipples are more frequently found in individuals with N-glycosylation defects.20
Regarding the limitations of this study, it is important to mention that it is based on clinical information present in the files of individuals with suspected CDG. Another important limitation regarding individuals from other regions of the country is that, after the release of the patient’s file to the services of origin, the authors have no information on the follow-up these individuals had, i.e., whether the patient completed the investigation or not.
ConclusionDespite the limitations of the study regarding the retrospective analysis of cases through the review of medical records and the lack of uniformity of care and diagnosis of patients from different regions of the country, it was observed that it is necessary to implement new diagnostic techniques for CDG, as well as a more comprehensive protocol for CDG investigation in the LEIM-HCPA. The findings show that although alterations in the TfIEF were reported in 51 cases, the CDG diagnosis was confirmed in only four individuals.
The obtained data also showed that the main symptoms that led to suspected CDG were neurological. However, individuals with inverted nipples, liver disease, hypoglycemia, ophthalmological alterations, and history of other cases in the family were the most likely to have altered TfIEF. Therefore, screening for CDG should be especially considered when these clinical manifestations are present.
FundingThis study received funding from Fundo de Incentivo à Pesquisa e Eventos do Hospital de Clínicas de Porto Alegre (FIPE-HCPA), funding number: 18-0324.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank their colleagues Zeniara Lompa, Régis Guidobono, Karen Lucas, and Tânia Braga, from the staff of the HCPA Medical Genetics Service, for their support of the study. They are also grateful for the financial support provided by FIPE-HCPA.
Please cite this article as: Magalhães AP, Burin MG, Souza CF, Bitencourt FH, Sebastião FM, Silva TO, et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten-year experience in a Brazilian center. J Pediatr (Rio J). 2020;96:710–6.
Study conducted at Universidade Federal do Rio Grande do Sul (UFRGS), Hospital de Clínicas de Porto Alegre (HCPA), Porto Alegre, RS, Brazil.




