



This study aimed to review the literature on the repercussions of the different inborn errors of immunity on growth, drawing attention to the diagnosis of this group of diseases in patients with growth disorders, as well as to enable the identification of the different causes of growth disorders in patients with inborn errors of immunity, which can help in its treatment.
Data sourcesNon‐systematic review of the literature, searching articles since 2000 in PubMed with the terms “growth”, “growth disorders”, “failure to thrive”, or “short stature” AND “immunologic deficiency syndromes”, “immune deficiency disease”, or “immune deficiency” NOT HIV. The Online Mendelian Inheritance in Man (OMIN) database was searched for immunodeficiencies and short stature or failure to thrive.
Data summaryInborn errors of immunity can affect growth in different ways, and some of them can change growth through multiple simultaneous mechanisms: genetic syndromes; disorders of the osteoarticular system; disorders of the endocrine system; reduction in caloric intake; catabolic processes; loss of nutrients; and inflammatory and/or infectious conditions.
ConclusionsThe type of inborn errors of immunity allows anticipating what type of growth disorder can be expected. The type of growth disorder can help in the diagnosis of clinical conditions related to inborn errors of immunity. In many inborn errors of immunity, the causes of poor growth are mixed, involving more than one factor. In many cases, impaired growth can be adjusted with proper inborn errors of immunity treatment or proper approach to the mechanism of growth impairment.
Revisão da literatura sobre as repercussões dos diferentes erros inatos da imunidade sobre o crescimento, chamar a atenção para o diagnóstico desse grupo de doenças em pacientes que apresentem desordens do crescimento, assim como permitir que se identifiquem as diferentes causas de alterações do crescimento em pacientes com erros inatos da imunidade, o que pode auxiliar em seu manejo.
Fonte dos dadosRevisão não sistemática da literatura, com busca de artigos desde 2000 no Pubmed com os termos “growth” ou “growth disorders” ou “failure to thrive” ou “short stature” AND “immunologic deficiency syndromes” ou “immune deficiency disease” ou “imune deficiency” NOT HIV. E buscas na base OMIN (Online Mendelian Inheritance in Man) por imunodeficiências e baixa estatura ou falha no crescimento (“failure to thrive”).
Síntese dos dadosHá diferentes modos pelos quais os erros inatos da imunidade podem afetar o crescimento e alguns deles podem alterar o crescimento por múltiplos mecanismos simultâneos: síndromes genéticas; afecções do aparelho osteoarticular; afecções do sistema endócrino; redução de aporte calórico; processos catabólicos: perda de nutrientes, assim como afecções inflamatórias e/ou infecciosas.
ConclusõesO tipo de erros inatos da imunidade permite prever que tipo de alteração no crescimento devemos esperar. O tipo de alteração no crescimento pode auxiliar no diagnóstico de condições clínicas associadas aos erros inatos da imunidade. Em muitos erros inatos da imunidade, as causas do crescimento deficiente são mistas, envolvem mais de um fator. Em muitos casos, o prejuízo do crescimento pode ser corrigido com o adequado tratamento dos erros inatos da imunidade ou adequada abordagem do mecanismo que causa o prejuízo do crescimento.
As imunodeficiências primárias (IDP) ou erros inatos da imunidade (EII), como mais recentemente tem se proposto denominar esse grupo de patologias, correspondem a um grupo bastante heterogêneo de doenças que afetam primariamente o sistema imune.1 As manifestações clínicas diferem sobremaneira dentro do grupo e envolvem quadros infecciosos, autoimunidade, inflamação, alergia e malignidades.2
Atualmente, há mais de 340 defeitos genéticos associados a imunodeficiência e desregulação imune. Causam doenças classificadas de acordo com o setor do sistema imune comprometido prioritariamente e as principais manifestações clínicas.1,2 A classificação dos EII1é composta por nove tabelas: 1 ‐ imunodeficiências que afetam imunidade celular e humoral, 2 ‐ imunodeficiências combinadas com características ou síndromes associadas, 3 ‐ defeitos predominantemente de anticorpos, 4 ‐ doenças de desregulação imune, 5 ‐ defeitos quantitativos ou funcionais de fagócitos, 6 ‐ defeitos da imunidade intrínseca e inata, 7 ‐ desordens autoinflamatórias, 8 ‐ deficiências do sistema complemento e 9 ‐ fenocópias de EII.
Os EII mais graves são os defeitos combinados de imunidade celular e humoral (tabela 1 da classificação) nos quais há acometimento da produção de anticorpos e do número de linfócitos. Nesse grupo, estão doenças associadas a quadros infecciosos graves por diversos tipos de agentes infecciosos (bactérias, fungos e vírus), denominadas como imunodeficiência combinada grave. Essa é considerada uma emergência médica, com prognóstico reservado caso não seja feito transplante de células‐tronco hematopoiéticas precocemente. Esses defeitos combinados podem estar associados a certas características clínicas ou síndromes (tabela 2 da classificação), tais como a síndrome de Wiskott‐Aldrich (eczema, plaquetopenia com plaquetas pequenas e infecções), a ataxia‐telangiectasia (ataxia do tipo cerebelar e telangiectasias óculo‐cutâneas), a síndrome velo‐cardio‐facial/DiGeorge (hipoparatireoidismo, cardiopatias conotruncais, insuficiência velo‐palatal, anormalidades faciais), as displasias imuno‐ósseas e a síndrome de Hiper‐IgE.
Os defeitos predominantemente de anticorpos (tabela 3 da classificação) representam aproximadamente 50% do total de EII. Estão classificadas nessa tabela a deficiência seletiva de IgA, agamaglobulinemia ligada ao X e a imunodeficiência comum variável.
As doenças de desregulação imune (tabela quatro da classificação) apresentam como principal característica manifestações de autoimunidade e/ou linfoproliferação. Exemplos de doenças dessa tabela são a síndrome linfoproliferativa autoimune (ALPS), a poliendocrinopatia com candidíase e a distrofia ectodérmica (Apeced), a desregulação imune com endocrinopatia e a enteropatia autoimune (Ipex) e Chédiak‐Higashi.
A tabela cinco da classificação engloba defeitos quantitativos (neutropenia e neutropenia cíclica) ou funcionais de fagócitos (deficiência de adesão leucocitária e doença granulomatosa crônica).
Os defeitos da imunidade inata (tabela 6 da classificação) envolvem quadros infecciosos recorrentes por vírus, fungos e/ou micobactérias.
Na tabela 7 da classificação encontramos um grupo de doenças em que há uma desregulação da imunidade inata e que são caracterizadas por processo inflamatório recorrente e/ou crônico, com ou sem febre, e não relacionado a autoimunidade ou infecções.
Defeitos do sistema do complemento (tabela 8 da classificação) estão relacionados a fenômenos autoimunes (particularmente lúpus eritematoso sistêmico) e infecções por germes encapsulados extracelulares, principalmente meningococos.
As fenocópias de EII (tabela 9 da classificação) são condições clínicas que se assemelham a algumas imunodeficiências de tabelas anteriores, mas que decorrem de mutações somáticas (mutações que acontecem durante o desenvolvimento do feto, em um determinado tipo celular, não são transmitidas à prole) ou autoanticorpos.
Falência do crescimento é observada em um grande número de condições clínicas.3 Mais comumente, está associada a reduzido aporte calórico por baixa ingesta, má absorção ou estados de hipercatabolismo, como em quadros infecciosos e inflamatórios. Outros mecanismos podem estar envolvidos, relacionados a alterações endócrinas, inclusive hipotireoidismo e deficiência de GH, ou displasias ósseas. Além disso, algumas síndromes genéticas e anormalidades cromossômicas podem levar a alterações do crescimento.3
Na dependência do defeito molecular e manifestações clínicas, os EII podem comprometer o crescimento por mecanismos diferentes e, em alguns casos, vários mecanismos simultaneamente. Portanto, esse grupo de doenças deve ser considerado no diagnóstico diferencial de baixa estatura e alterações no crescimento.
O reconhecimento e tratamento precoces dos EII melhoram seu prognóstico. Por outro lado, conhecer os mecanismos pelos quais o crescimento pode ser prejudicado nesse grupo de doenças possibilita tratamento específico que melhore o crescimento pondero‐estatural dos pacientes.
ObjetivosFazer uma revisão da literatura sobre as repercussões dos diferentes EII sobre o crescimento, de maneira a chamar a atenção para o diagnóstico desse grupo de doenças em pacientes que apresentem desordens do crescimento, assim como permitir que se identifiquem as diferentes causas de alterações do crescimento em pacientes com EII.
MétodosFoi feita uma revisão não sistemática da literatura, buscaram‐se artigos publicados nos últimos 18 anos (desde 2000) na base de dados Pubmed com diferentes combinações dos termos “growth” ou “growth disorders” ou “failure to thrive” ou “short stature” AND “immunologic deficiency syndromes” ou “immune deficiency disease” ou “imune deficiency” NOT HIV. Usamos filtros de artigos de revisão, em inglês ou francês. Também fizemos buscas na base OMIN (Online Mendelian Inheritance in Man) por imunodeficiências e baixa estatura ou falha no crescimento (“failure to thrive”).
O crescimento pondero‐estatural é avaliado por medidas de peso, altura/comprimento, perímetro cefálico e índice de massa corporal (IMC), inseridas em gráficos da OMS e CDC.4 Outras medidas relacionadas ao crescimento são a avaliação de proporção corporal, de maturidade óssea e de desenvolvimento dentário.4 Neste texto, vamos analisar alterações relacionadas aos EII que envolvem essas medidas, exceto IMC e perímetro cefálico.
ResultadosO crescimento é um processo complexo no qual diversos fatores genéticos e ambientais podem interferir.5 Assim, o crescimento do indivíduo depende de um somatório de condições para que ocorra de forma adequada e atinja a sua plenitude. Entre esses fatores podemos destacar a oferta adequada de nutrientes, a capacidade de absorção desses nutrientes, o potencial genético herdado e a integridade das vias endócrinas e osteoarticular. Outro ponto fundamental é que o organismo tenha o equilíbrio entre as fontes energéticas e o gasto calórico.
Doenças genéticas podem afetar a função hormonal ou o sistema osteoarticular. Alterações adquiridas do crescimento se relacionam a fatores psicossociais e/ou diferentes doenças.3 Causas adquiridas de crescimento insuficiente estão relacionadas a alterações endócrinas, baixo aporte calórico, má absorção e aumento do gasto calórico (tal como em processos infecciosos, inflamatórios ou neoplásicos).
Em linhas gerais, nas crianças com doenças crônicas, a falência do crescimento está relacionada aos efeitos da nutrição inadequada e ao dispêndio energético resultante do processo inflamatório gerado pela própria doença. Um estado de subnutrição crônica e a liberação de citocinas inflamatórias são determinantes para a resistência ao hormônio do crescimento (GH).6 Citocinas pró‐inflamatórias, como o Fator de Necrose Tumoral alfa (TNF‐α), agem sobre o sistema nervoso central, alteram as vias do apetite e do metabolismo energético e levam à perda muscular.7
O eixo GH/IGF1 (insulin‐like growth factor‐1) tem papel fundamental no crescimento. Alterações no metabolismo e resistência dos órgãos ao GH têm sido descritos como um dos principais fatores que contribuem para o retardo de crescimento de pacientes com doença inflamatória intestinal na infância (situação comum entre os pacientes com EII).8 Múltiplas citocinas presentes nos processos inflamatórios (de natureza autoimune ou infecciosa) inibem as vias que envolvem IGF‐1. O aumento de IL6, presente de forma generalizada nos quadros de inflamação crônica, parece representar um dos principais mecanismos que afetam o desenvolvimento esquelético.9
Nas imunodeficiências primárias, a grande maioria das crianças apresenta um número aumentado de infecções ou infecções de maior gravidade, que demandam uma resposta inflamatória persistente ou recorrente, estimulam um grande número de citocinas de forma reacional. Da mesma forma, uma outra parcela dos EII apresenta alterações na regulação do sistema imune, que se associa a uma redução ou ausência dos mecanismos de controle do próprio sistema imunológico e resulta em um processo inflamatório crônico, de intensidade variável de acordo com o defeito imune específico. Além disso, a presença de doença inflamatória intestinal nessas crianças com IDP ainda promove redução da absorção dos nutrientes, o que agrava o quadro.
Além da relação processo inflamatório‐crescimento‐nutrição, muitos pacientes com EII apresentam síndromes genéticas associadas à baixa estatura, como cromossomopatias, defeitos de reparo do DNA e displasia osteoarticular. Além disso, muitos EII podem estar associados a doenças do sistema endócrino que alteram níveis de hormônios essenciais ao crescimento normal da criança e produzem falência do crescimento.
Em resumo, há diferentes modos pelos quais os EII podem afetar o crescimento e alguns desses defeitos da imunidade podem alterar o crescimento por múltiplos mecanismos simultâneos. Podemos dividi‐los da seguinte forma:
- ‐
síndromes genéticas;
- ‐
afecções do aparelho osteoarticular;
- ‐
afecções do sistema endócrino;
- ‐
redução de aporte calórico;
- ‐
processos catabólicos.
O diagnóstico precoce dos EII tem relevantes implicações prognósticas. No mundo todo, têm‐se divulgado 10 sinais de alerta desenvolvidos pela Fundação Jeffrey Modell, que têm por base a opinião de especialistas (tabela 1). Apesar de serem muito usados, não há estudos que confirmem sua eficácia na prática clínica.10 História familiar de imunodeficiência combinada com uso de antibióticos venosos, infecções profundas, falha no crescimento, história de morte precoce de irmão e consanguinidade entre os pais foram definidos como melhores preditores de EII na criança segundo alguns estudos.10,11
Sinais de alerta para EII da Fundação Jeffrey Modell adaptados para o nosso meio
| Duas ou mais pneumonias no último ano |
| Quatro ou mais otites no último ano |
| Estomatites de repetição ou monilíase por mais de dois meses |
| Abscessos de repetição ou ectima |
| Um episódio de infecção sistêmica grave (meningite, osteoartrite, septicemia) |
| Infecções intestinais de repetição/diarreia crônica |
| Asma grave, doença do colágeno ou doença autoimune |
| Efeito adverso ao BCG e/ou infecção por micobactéria |
| Fenótipo clínico sugestivo de síndrome associada a imunodeficiência |
| História familiar de imunodeficiência |
Fonte: http://www.bragid.org.br/.
A investigação inicial dos EII é composta de hemograma completo, dosagem de imunoglobulinas (A, M, G e E), contagem de subpopulações de linfócitos (CD3, CD4, CD8, CD19 e CD56/16), dosagem de CH50 (atividade hemolítica total do complemento) e DHR (avaliação do burst oxidativo de neutrófilos).11
A análise das curvas de crescimento pode auxiliar na investigação dos EII e dos mecanismos pelos quais o crescimento é afetado. É relevante distinguir se o prejuízo do crescimento já se encontra presente ao nascimento, por conta de crescimento intrauterino retardado, ou se o paciente é eutrófico na infância precoce e apresenta prejuízo do crescimento mais tarde, assim como é relevante observar a relação entre curvas de peso e comprimento/altura, além de proporção corporal.5,12
Alterações de crescimento por doenças genéticas (osteoarticulares ou cromossomopatias) afetam as curvas desde o nascimento: o paciente nasce pequeno e se mantém abaixo das curvas ao longo da infância.5,12 Baixa estatura com alteração de proporção corporal entre tronco e membros está, em geral, relacionada a displasia óssea.13
Nas desordens endócrinas, a altura é afetada antes ou simultaneamente ao peso e a relação peso x altura encontra‐se normal ou aumentada. Em defeitos nutricionais (baixo aporte, alteração de absorção ou catabolismo) o peso é afetado antes da altura e a relação peso x altura é baixa.3 Nessas desordens, habitualmente encontramos atraso da idade óssea.
A seguir, analisamos cada um dos mecanismos envolvidos na falência do crescimento em pacientes com EII, em separado:
Síndromes genéticas (com ou sem alterações do sistema osteoarticular)Crescimento intrauterino retardado está comumente associado a EII associados a cromossomopatias, displasias ósseas (alterações da formação óssea) e defeitos no reparo de DNA. Nesses últimos, os pacientes também costumam apresentar microcefalia ao nascimento.14
Há uma grande quantidade de síndromes com defeitos do sistema imune associados à baixa estatura sem alteração de proporção corporal (tabela 2).13 Várias doenças cromossômicas estão associadas a EII, particularmente com defeitos na produção de anticorpos.15
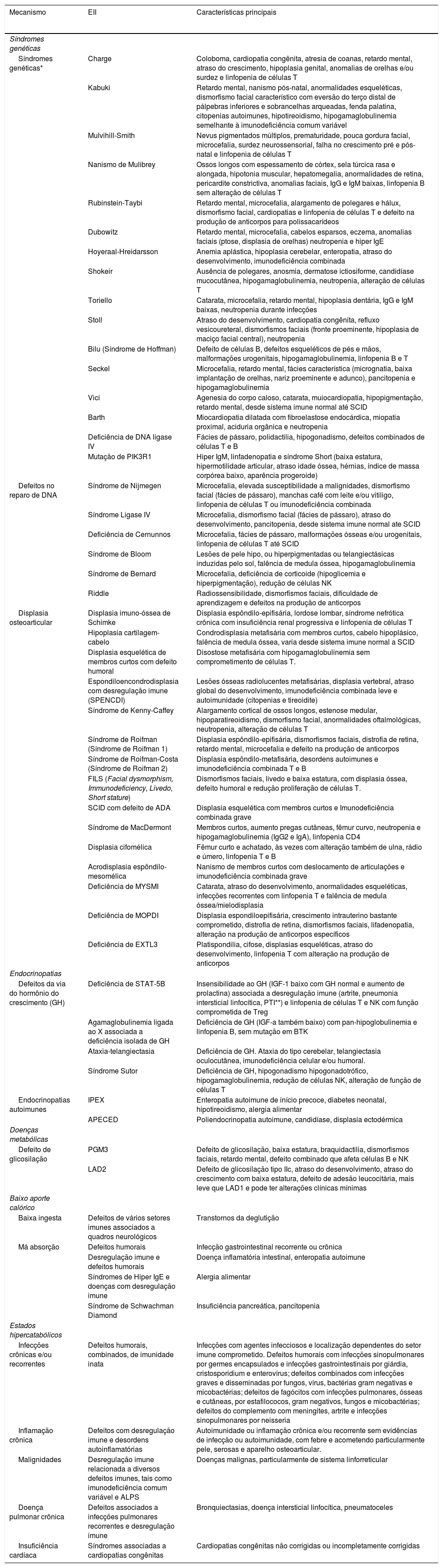
EII e baixa estatura – mecanismos, EII e características principais
| Mecanismo | EII | Características principais |
|---|---|---|
| Síndromes genéticas | ||
| Síndromes genéticas* | Charge | Coloboma, cardiopatia congênita, atresia de coanas, retardo mental, atraso do crescimento, hipoplasia genital, anomalias de orelhas e/ou surdez e linfopenia de células T |
| Kabuki | Retardo mental, nanismo pós-natal, anormalidades esqueléticas, dismorfismo facial característico com eversão do terço distal de pálpebras inferiores e sobrancelhas arqueadas, fenda palatina, citopenias autoimunes, hipotireoidismo, hipogamaglobulinemia semelhante à imunodeficiência comum variável | |
| Mulvihill-Smith | Nevus pigmentados múltiplos, prematuridade, pouca gordura facial, microcefalia, surdez neurossensorial, falha no crescimento pré e pós-natal e linfopenia de células T | |
| Nanismo de Mulibrey | Ossos longos com espessamento de córtex, sela túrcica rasa e alongada, hipotonia muscular, hepatomegalia, anormalidades de retina, pericardite constrictiva, anomalias faciais, IgG e IgM baixas, linfopenia B sem alteração de células T | |
| Rubinstein-Taybi | Retardo mental, microcefalia, alargamento de polegares e hálux, dismorfismo facial, cardiopatias e linfopenia de células T e defeito na produção de anticorpos para polissacarídeos | |
| Dubowitz | Retardo mental, microcefalia, cabelos esparsos, eczema, anomalias faciais (ptose, displasia de orelhas) neutropenia e hiper IgE | |
| Hoyeraal-Hreidarsson | Anemia aplástica, hipoplasia cerebelar, enteropatia, atraso do desenvolvimento, imunodeficiência combinada | |
| Shokeir | Ausência de polegares, anosmia, dermatose ictiosiforme, candidíase mucocutânea, hipogamaglobulinemia, neutropenia, alteração de células T | |
| Toriello | Catarata, microcefalia, retardo mental, hipoplasia dentária, IgG e IgM baixas, neutropenia durante infecções | |
| Stoll | Atraso do desenvolvimento, cardiopatia congênita, refluxo vesicoureteral, dismorfismos faciais (fronte proeminente, hipoplasia de maciço facial central), neutropenia | |
| Bilu (Síndrome de Hoffman) | Defeito de células B, defeitos esqueléticos de pés e mãos, malformações urogenitais, hipogamaglobulinemia, linfopenia B e T | |
| Seckel | Microcefalia, retardo mental, fácies característica (micrognatia, baixa implantação de orelhas, nariz proeminente e adunco), pancitopenia e hipogamaglobulinemia | |
| Vici | Agenesia do corpo caloso, catarata, muiocardiopatia, hipopigmentação, retardo mental, desde sistema imune normal até SCID | |
| Barth | Miocardiopatia dilatada com fibroelastose endocárdica, miopatia proximal, aciduria orgânica e neutropenia | |
| Deficiência de DNA ligase IV | Fácies de pássaro, polidactilia, hipogonadismo, defeitos combinados de células T e B | |
| Mutação de PIK3R1 | Hiper IgM, linfadenopatia e síndrome Short (baixa estatura, hipermotilidade articular, atraso idade óssea, hérnias, índice de massa corpórea baixo, aparência progeroide) | |
| Defeitos no reparo de DNA | Síndrome de Nijmegen | Microcefalia, elevada susceptibilidade a malignidades, dismorfismo facial (fácies de pássaro), manchas café com leite e/ou vitiligo, linfopenia de células T ou imunodeficiência combinada |
| Síndrome Ligase IV | Microcefalia, dismorfismo facial (fácies de pássaro), atraso do desenvolvimento, pancitopenia, desde sistema imune normal ate SCID | |
| Deficiência de Cernunnos | Microcefalia, fácies de pássaro, malformações ósseas e/ou urogenitais, linfopenia de células T até SCID | |
| Síndrome de Bloom | Lesões de pele hipo, ou hiperpigmentadas ou telangiectásicas induzidas pelo sol, falência de medula óssea, hipogamaglobulinemia | |
| Síndrome de Bernard | Microcefalia, deficiência de corticoide (hipoglicemia e hiperpigmentação), redução de células NK | |
| Riddle | Radiossensibilidade, dismorfismos faciais, dificuldade de aprendizagem e defeitos na produção de anticorpos | |
| Displasia osteoarticular | Displasia imuno-óssea de Schimke | Displasia espôndilo-epifisária, lordose lombar, síndrome nefrótica crônica com insuficiência renal progressiva e linfopenia de células T |
| Hipoplasia cartilagem-cabelo | Condrodisplasia metafisária com membros curtos, cabelo hipoplásico, falência de medula óssea, varia desde sistema imune normal a SCID | |
| Displasia esquelética de membros curtos com defeito humoral | Disostose metafisária com hipogamaglobulinemia sem comprometimento de células T. | |
| Espondiloencondrodisplasia com desregulação imune (SPENCDI) | Lesões ósseas radiolucentes metafisárias, displasia vertebral, atraso global do desenvolvimento, imunodeficiência combinada leve e autoimunidade (citopenias e tireoidite) | |
| Síndrome de Kenny-Caffey | Alargamento cortical de ossos longos, estenose medular, hipoparatireoidismo, dismorfismo facial, anormalidades oftalmológicas, neutropenia, alteração de células T | |
| Síndrome de Roifman (Síndrome de Roifman 1) | Displasia espôndilo-epifisária, dismorfismos faciais, distrofia de retina, retardo mental, microcefalia e defeito na produção de anticorpos | |
| Síndrome de Roifman-Costa (Síndrome de Roifman 2) | Displasia espôndilo-metafisária, desordens autoimunes e imunodeficiência combinada T e B | |
| FILS (Facial dysmorphism, Immunodeficiency, Livedo, Short stature) | Dismorfismos faciais, livedo e baixa estatura, com displasia óssea, defeito humoral e redução proliferação de células T. | |
| SCID com defeito de ADA | Displasia esquelética com membros curtos e Imunodeficiência combinada grave | |
| Síndrome de MacDermont | Membros curtos, aumento pregas cutâneas, fêmur curvo, neutropenia e hipogamaglobulinemia (IgG2 e IgA), linfopenia CD4 | |
| Displasia cifomélica | Fêmur curto e achatado, às vezes com alteração também de ulna, rádio e úmero, linfopenia T e B | |
| Acrodisplasia espôndilo-mesomélica | Nanismo de membros curtos com deslocamento de articulações e imunodeficiência combinada grave | |
| Deficiência de MYSMI | Catarata, atraso do desenvolvimento, anormalidades esqueléticas, infecções recorrentes com linfopenia T e falência de medula óssea/mielodisplasia | |
| Deficiência de MOPDI | Displasia espondiloepifisária, crescimento intrauterino bastante comprometido, distrofia de retina, dismorfismos faciais, lifadenopatia, alteração na produção de anticorpos específicos | |
| Deficiência de EXTL3 | Platispondilia, cifose, displasias esqueléticas, atraso do desenvolvimento, linfopenia T com alteração na produção de anticorpos | |
| Endocrinopatias | ||
| Defeitos da via do hormônio do crescimento (GH) | Deficiência de STAT-5B | Insensibilidade ao GH (IGF-1 baixo com GH normal e aumento de prolactina) associada a desregulação imune (artrite, pneumonia intersticial linfocítica, PTI**) e linfopenia de células T e NK com função comprometida de Treg |
| Agamaglobulinemia ligada ao X associada a deficiência isolada de GH | Deficiência de GH (IGF-a também baixo) com pan-hipoglobulinemia e linfopenia B, sem mutação em BTK | |
| Ataxia-telangiectasia | Deficiência de GH. Ataxia do tipo cerebelar, telangiectasia oculocutânea, imunodeficiência celular e/ou humoral. | |
| Síndrome Sutor | Deficiência de GH, hipogonadismo hipogonadotrófico, hipogamaglobulinemia, redução de células NK, alteração de função de células T | |
| Endocrinopatias autoimunes | IPEX | Enteropatia autoimune de início precoce, diabetes neonatal, hipotireoidismo, alergia alimentar |
| APECED | Poliendocrinopatia autoimune, candidíase, displasia ectodérmica | |
| Doenças metabólicas | ||
| Defeito de glicosilação | PGM3 | Defeito de glicosilação, baixa estatura, braquidactilia, dismorfismos faciais, retardo mental, defeito combinado que afeta células B e NK |
| LAD2 | Defeito de glicosilação tipo IIc, atraso do desenvolvimento, atraso do crescimento com baixa estatura, defeito de adesão leucocitária, mais leve que LAD1 e pode ter alterações clínicas mínimas | |
| Baixo aporte calórico | ||
| Baixa ingesta | Defeitos de vários setores imunes associados a quadros neurológicos | Transtornos da deglutição |
| Má absorção | Defeitos humorais | Infecção gastrointestinal recorrente ou crônica |
| Desregulação imune e defeitos humorais | Doença inflamatória intestinal, enteropatia autoimune | |
| Síndromes de Hiper IgE e doenças com desregulação imune | Alergia alimentar | |
| Síndrome de Schwachman Diamond | Insuficiência pancreática, pancitopenia | |
| Estados hipercatabólicos | ||
| Infecções crônicas e/ou recorrentes | Defeitos humorais, combinados, de imunidade inata | Infecções com agentes infecciosos e localização dependentes do setor imune comprometido. Defeitos humorais com infecções sinopulmonares por germes encapsulados e infecções gastrointestinais por giárdia, cristosporidium e enterovírus; defeitos combinados com infecções graves e disseminadas por fungos, vírus, bactérias gram negativas e micobactérias; defeitos de fagócitos com infecções pulmonares, ósseas e cutâneas, por estafilococos, gram negativos, fungos e micobactérias; defeitos do complemento com meningites, artrite e infecções sinopulmonares por neisseria |
| Inflamação crônica | Defeitos com desregulação imune e desordens autoinflamatórias | Autoimunidade ou inflamação crônica e/ou recorrente sem evidências de infecção ou autoimunidade, com febre e acometendo particularmente pele, serosas e aparelho osteoarticular. |
| Malignidades | Desregulação imune relacionada a diversos defeitos imunes, tais como imunodeficiência comum variável e ALPS | Doenças malignas, particularmente de sistema linforreticular |
| Doença pulmonar crônica | Defeitos associados a infecções pulmonares recorrentes e desregulação imune | Bronquiectasias, doença intersticial linfocítica, pneumatoceles |
| Insuficiência cardíaca | Síndromes associadas a cardiopatias congênitas | Cardiopatias congênitas não corrigidas ou incompletamente corrigidas |
A maioria das síndromes genéticas, com ou sem acometimento osteoarticular, encontra‐se listada nas tabelas 1 e 2 da classificação dos EII, que incluem os defeitos combinados de células T e B e defeitos combinados associados a síndromes, respectivamente.1
Defeitos genéticos em proteínas envolvidas no reparo de DNA costumam estar associados a anormalidades imunológicas, que variam desde um acometimento grave, com fenótipo de imunodeficiência combinada grave (como é o caso da deficiência de ligase IV e Cernunnos) até defeitos mais leves. Defeitos no complexo GINS, essencial para a reprodução do DNA antes da divisão celular, afetam particularmente neutrófilos e células NK, produzem fenótipo diferente da imunodeficiência combinada.16–18
Afecções do aparelho osteoarticularDisplasias ósseas afetam osso e cartilagem de crescimento e apresentam achados radiológicos específicos, dependem do defeito genético (tabela 2) e podem produzir, além de prejuízo do crescimento, alterações da proporção corporal e deformidades.13
As displasias associadas a alterações do sistema imune são denominadas displasias imuno‐ósseas e estão relacionadas a níveis variáveis de deficiência de células T e/ou B. Há relatos de hipocondroplasia (alterações esqueléticas menos graves do que na acondroplasia) e outros defeitos imunológicos, como linfopenia CD4 e deficiência de IgA.19
Pacientes com hipoplasia cartilagem cabelo manifestam baixa estatura grave, com membros curtos, displasia ectodérmica, anemia, imunodeficiência variável (em geral combinada, de início mais tardio) e maior susceptibilidade a doenças malignas.20 Os achados radiológicos são bem variáveis, mas caracteristicamente apresentam ossos curtos e alargados, com metáfises bojudas e irregulares e epífises globulares em joelhos e tornozelos.21
Outras displasias imuno‐ósseas são a displasia esquelética de membros curtos com imunodeficiência combinada, Síndrome de MacDermot, displasia cifomélica, acrodisplasia espondilomesomélica, displasia esquelética de membros curtos com imunodeficiência humoral, displasia de Schimke, síndrome de Roifman, síndrome SPENCDI, síndrome de Kenny‐Caffey, deficiência de MYSMI, deficiência de MOPDI e deficiência de EXTL3 1,13 (tabela 2).
Na imunodeficiência combinada por deficiência de ADA, há relato de baixa estatura com membros curtos e deformidades de gradil costal que podem, ao menos em parte, ser reversíveis com o tratamento de reposição enzimática, transplante de medula ou terapia gênica.22
Quadros infecciosos (mais comuns em defeitos dos fagócitos) ou inflamatórios (doenças autoinflamatórias) também podem causar crescimento assimétrico de membros com alteração de proporções corporais e/ou deformidades localizadas.23
Afecções do sistema endócrinoAs alterações endócrinas relacionadas aos EII podem ser de natureza autoimune ou ser relacionadas a alterações da via do hormônio do crescimento (GH). Nesse segundo caso, estão o defeito de STAT5b e agamaglobulinemia com defeito de GH.
EII caracterizados por autoimunidade ou desregulação imune causam impacto no crescimento por conta de endocrinopatias secundárias. Doenças endócrinas autoimunes, particularmente de tireoide, paratireoide e diabetes mellitus estão em sua maioria na tabela 4 da classificação de EII, a de desregulação imune.1
No defeito de STAT5b há comprometimento da produção do fator de crescimento semelhante à insulina 1 (IGF1), assemelha‐se fenotipicamente à síndrome de insensibilidade ao GH (síndrome de Laron).24 Mais recentemente, mutações monoalélicas dominantes negativas foram descritas.24,25 Há redução das dosagens de IGF1, IGFBP3 (proteína 3 ligadora de IGF) e ALS e sensível aumento de prolactina (subunidade ácido lábil de IGFB3).24,26 Esse defeito está também associado a eczema, doença pulmonar crônica (pneumonia linfocítica, fibrose), hipergamaglobulinemia e linfopenia de células T e Treg.27
Pacientes com mutações com ganho de função de STAT3 também podem apresentar baixa estatura, com mecanismos não completamente esclarecidos.28 É possível que ocorra por ativação de STAT5 e insensibilidade parcial a GH.29,30 Entretanto, esses pacientes apresentam múltiplas manifestações precoces de autoimunidade que requerem tratamento imunossupressor, o que torna complicado diferenciar da baixa estatura pelo uso crônico/recorrentes de corticoides sistêmicos.
Outros defeitos na sinalização de citocinas que podem se manifestar com baixa estatura são os da via da PI3K.31,32 A sinalização alterada na via PI3K‐AKT‐mTOR pode levar a resistência a insulina e fator de crescimento, com prejuízo na divisão celular e consequente atraso no crescimento.33,34 Os pacientes com mutação de PI3KR1 podem se apresentar com a síndrome Short (baixa estatura, hiperextensibilidade articular, atraso da dentição, lipodistrofia parcial), além de hiper IgM e linfadenopatia.29,30
A agamaglobulinemia e o defeito isolado de GH ligada ao X apresentam muitas semelhanças com a agamagobulinemia ligada ao X, com pan‐hipogmaglobulinemia e baixos níveis de linfócitos B, mas não há mutação ou alteração de expressão de BTK.35,36
Redução na secreção central de GH está descrita também na ataxia‐telangiectasia.37
Redução do aporte calóricoAlterações do aporte calórico podem acontecer por simples baixa ingesta de nutrientes ou por transtornos da deglutição em EII que evoluem com alteração neurológica motora, tal como na ataxia‐telangiectasia.1
Em outros EII, pode haver significativa má absorção de nutrientes relacionada a processo inflamatório do trato digestivo (doença inflamatória), de natureza infecciosa, alérgica ou ainda por disfunção pancreática.1
Processos catabólicosEstão envolvidos em doenças de praticamente todas as tabelas da classificação dos EII, em maior ou menor grau.
Alterações ponderais agudas estão particularmente relacionas aos quadros infecciosos e/ou inflamatórios agudos e a malignidades. Habitualmente, acontece recuperação das curvas uma vez que o processo esteja controlado.
Entretanto, quadros inflamatórios/infecciosos crônicos, assim como perdas gastrointestinais principalmente, causam prejuízo mais persistente das curvas de crescimento. Podem contribuir para estabelecimento de um estado hipercatabólico, malignidades, doença pulmonar crônica e insuficiência cardíaca.
Em boa parte dos EII, há mais de um fator que contribui para que haja um prejuízo do crescimento. Um exemplo dessa situação seria o IPEX, no qual doenças endócrinas (hipotireoidismo autoimune, diabetes I), perda de nutrientes (enteropatia autoimune) e baixo aporte de nutrientes (alergia alimentar) contribuem para prejudicar o crescimento.
No tabela 2 há um resumo dos mecanismos envolvidos na falha do crescimento em diversos EII e suas características principais.
Alterações especificamente do desenvolvimento dentário podem ocorrer em alguns EII. São descritos principalmente hipoplasia dentária na displasia ectodérmica com imunodeficiência (NEMO e outros), retardo da troca de dentes na síndrome de hiper IgE autossômica dominante e queda precoce de dentes da neutropenia cíclica.1
Alterações de maturidade óssea são bem descritas em endocrinopatias de tireoide e/ou paratireoide, associadas principalmente a EII com desregulação imune. Doenças crônicas de qualquer tipo, inclusive quadros infecciosos/inflamatórios típicos de EII, costumam promover atraso da maturação óssea avaliada pela idade óssea.38 Alteração da produção de IGF1 (Insulin‐like Growth Factor 1) acontece em desnutrição, doença inflamatória intestinal e doenças hepáticas,5 que podem fazer parte do quadro clínico de muitos EII.
Uma vez confirmado o diagnóstico de EII, o aporte nutricional adequado, inclusive a avaliação de necessidade de suplementação individualizada para cada paciente, o controle das infecções e do processo inflamatório e a monitoração e o tratamento daqueles pacientes portadores de síndromes associadas a alterações endócrinas são importantes para manter o crescimento dos pacientes com EII da melhor forma. Os pacientes com alterações sindrômicas associadas ao aparelho osteoarticular devem ser reconhecidos de forma precoce para medidas ortopédico‐fisioterápicas sejam instituídas e reduzam ao máximo o impacto dessas malformações.39
ConclusãoDe uma forma geral, os pacientes com EII apresentam um risco maior de falência do crescimento. O tipo de EII permite prever que tipo de alteração no crescimento devemos esperar. Por outro lado, o tipo de alteração no crescimento pode auxiliar no diagnóstico de condições clínicas associadas aos EII. Entretanto, em muitos EII, as causas do crescimento deficiente são mistas, envolvem mais de um fator.
Em pacientes que crescem abaixo das curvas desde o nascimento, devemos cogitar síndromes genéticas associadas a defeitos do sistema imune que envolvam ou não alterações osteoarticulares.
Alterações pós‐natais de curvas de crescimento, nas quais há prejuízo inicial da altura, devem nos fazer considerar desordens endócrinas associadas a EII ou ainda doenças osteoarticulares. Nessas últimas é importante estar atento para alterações de proporção corporal ou deformidades (fig. 1).

Ainda hoje, a falta de reconhecimento precoce dos EII leva a um diagnóstico tardio, privam‐se os pacientes de tratamento adequado precocemente, com consequências indesejáveis inclusive no crescimento de crianças e adolescentes. É fundamental estar atento aos sinais de alerta para EII diante de alterações do crescimento (tabela 3).
Sinais de alerta para EII diante de alterações do crescimento
| História familiar de EII |
|---|
| Infecções recorrentes e/ou graves e/ou pouco responsivas ao tratamento e/ou por germes oportunistas |
| Uso de antibiótico venoso para sepse |
| Duas ou mais endocrinopatias autoimunes |
| Endocrinopatia autoimune precoce |
| Manifestações de autoimunidade múltiplas e/ou precoces |
| Febre recorrente e/ou persistente |
| Alergia grave, com difícil controle |
| Diarreia persistente com ou sem infecção |
| Fenótipo clínico sugestivo de síndromes associadas a EII |
| Morte precoce de irmão |
| Hipogamaglobulinemia na eletroforese de proteínas |
| Linfopenia no hemograma |
O diagnóstico e o tratamento adequados e precoces, com equipe multidisciplinar (nutrição, fisioterapia), são importantes para mantermos, dentro do possível, o crescimento adequado dos pacientes. Além disso, em muitos casos, o prejuízo do crescimento pode ser corrigido com o adequado tratamento do EII.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Goudouris ES, Segundo GR, Poli C. Repercussions of inborn errors of immunity on growth. J Pediatr. 2019;95:S49–S58.




