



This study aimed to review the literature on the repercussions of the different inborn errors of immunity on growth, drawing attention to the diagnosis of this group of diseases in patients with growth disorders, as well as to enable the identification of the different causes of growth disorders in patients with inborn errors of immunity, which can help in their treatment.
Data sourcesNon-systematic review of the literature, searching articles since 2000 in PubMed with the terms “growth”, “growth disorders”, “failure to thrive”, or “short stature” AND “immunologic deficiency syndromes”, “immune deficiency disease”, or “immune deficiency” NOT HIV. The Online Mendelian Inheritance in Man (OMIN) database was searched for immunodeficiencies and short stature or failure to thrive.
Data summaryInborn errors of immunity can affect growth in different ways, and some of them can change growth through multiple simultaneous mechanisms: genetic syndromes; disorders of the osteoarticular system; disorders of the endocrine system; reduction in caloric intake; catabolic processes; loss of nutrients; and inflammatory and/or infectious conditions.
ConclusionsThe type of inborn errors of immunity allows anticipating what type of growth disorder can be expected. The type of growth disorder can help in the diagnosis of clinical conditions related to inborn errors of immunity. In many inborn errors of immunity, the causes of poor growth are mixed, involving more than one factor. In many cases, impaired growth can be adjusted with proper inborn errors of immunity treatment or proper approach to the mechanism of growth impairment.
Revisão da literatura sobre as repercussões dos diferentes erros inatos da imunidade sobre o crescimento, chamar a atenção para o diagnóstico desse grupo de doenças em pacientes que apresentem desordens do crescimento, assim como permitir que se identifiquem as diferentes causas de alterações do crescimento em pacientes com erros inatos da imunidade, o que pode auxiliar em seu manejo.
Fonte dos dadosRevisão não sistemática da literatura, com busca de artigos desde 2000 no Pubmed com os termos “growth” ou “growth disorders” ou “failure to thrive” ou “short stature” AND “immunologic deficiency syndromes” ou “immune deficiency disease” ou “imune deficiency” NOT HIV. E buscas na base OMIN (Online Mendelian Inheritance in Man) por imunodeficiências e baixa estatura ou falha no crescimento (“failure to thrive”).
Síntese dos dadosHá diferentes modos pelos quais os erros inatos da imunidade podem afetar o crescimento e alguns deles podem alterar o crescimento por múltiplos mecanismos simultâneos: síndromes genéticas; afecções do aparelho osteoarticular; afecções do sistema endócrino; redução de aporte calórico; processos catabólicos: perda de nutrientes, assim como afecções inflamatórias e/ou infecciosas.
ConclusõesO tipo de erros inatos da imunidade permite prever que tipo de alteração no crescimento devemos esperar. O tipo de alteração no crescimento pode auxiliar no diagnóstico de condições clínicas associadas aos erros inatos da imunidade. Em muitos erros inatos da imunidade, as causas do crescimento deficiente são mistas, envolvem mais de um fator. Em muitos casos, o prejuízo do crescimento pode ser corrigido com o adequado tratamento dos erros inatos da imunidade ou adequada abordagem do mecanismo que causa o prejuízo do crescimento.
Primary immunodeficiencies (PID) or inborn errors of immunity (IEI), the term recently proposed to referrer to this group of pathologies, correspond to a quite heterogeneous group of diseases primarily affecting the immune system.1 The clinical manifestations differ greatly within the group and involve infectious conditions, autoimmunity, inflammation, allergy, and malignancies.2
Currently, there are over 340 genetic defects related to immunodeficiency and immune dysregulation; they cause diseases that are classified according to the sector of immune system that is primarily impaired as well as the main clinical manifestations.1,2 IEI classification1 is composed of nine tables: 1 – immunodeficiencies affecting cellular and humoral immunity; 2 – combined immunodeficiencies with associated or syndromic features; 3 – predominantly antibody deficiencies; 4 – diseases of immune dysregulation; 5 – congenital defects of phagocyte number or function ; 6 – defects in intrinsic and innate immunity disorders; 7 – autoinflammatory disorders; 8 – complement deficiencies; and 9 – phenotypes of inborn errors of immunity.
The most severe IEI are the combined cellular and humoral immune defects (Table 1 of the classification), in which there is impaired production of antibodies and number of lymphocytes. This group comprises diseases associated with severe infectious conditions caused by several types of infectious agents (bacteria, fungi, and virus), termed severe combined immunodeficiency. It is deemed a medical emergency, with poor prognosis if hematopoietic stem cell transplantation is not performed early. These combined defects can be associated with certain clinical characteristics or syndromes (Table 2 of the classification), such as Wiskott-Aldrich syndrome (eczema, small-platelet thrombocytopenia, and infections), ataxia-telangiectasia (cerebellar-type ataxia and oculocutaneous telangiectasias), velo-cardio-facial/DiGeorge syndrome (hypoparathyroidism, conotruncal heart diseases, velopalatal insufficiency, facial abnormalities); immuno-osseous dysplasia; and hyper-IgE syndromes.
Predominantly antibody defects (Table 3 of the classification) represent approximately 50% of the total IEI. Selective IgA deficiency, X-linked agammaglobulinemia, and common variable immunodeficiency are classified in this table.
In immune dysregulation diseases (Table 4 of the classification), autoimmunity and/or lymphoproliferation conditions are the main characteristics. Examples of the diseases in this table include autoimmune lymphoproliferative syndrome (ALPS), autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy (APECED), immune dysregulation with autoimmune endocrinopathy and autoimmune enteropathy (IPEX), and Chédiak-Higashi syndrome.
Table 5 of the classification comprises quantitative (neutropenia and cyclic neutropenia) or functional phagocyte defects (leukocyte adhesion deficiency and chronic granulomatous disease).
Defects in intrinsic and innate immunity (Table 6 of the classification) include recurrent infections by virus, fungi, and/or mycobacteria.
Table 7 of the classification presents a group of diseases with innateimmunity dysregulation, characterized by a recurring and/or chronic inflammatory process, with or without fever, and not associated with autoimmunity or infections.
Deficiencies in the complement system (Table 8 of the classification) are associated with autoimmune conditions (especially systemic lupus erythematosus) and infections caused by extracellular encapsulated bacteria, mainly meningococci.
IEI phenocopies (Table 9 of the classification) are clinical conditions similar to some immunodeficiencies described in previoutables; however, they arise from somatic mutations (mutations happening while the fetus is developing, in a certain cell type, not transmitted to offspring) or autoantibodies.
Growth failure is observed in a large number of clinical conditions.3 It is usually associated with reduced caloric intake due to low ingestion, malabsorption, or hypercatabolic states, as in infectious and inflammatory conditions. Other mechanisms associated with bone dysplasias or endocrine disorders can be involved, including hypothyroidism and growth hormone (GH) deficiency. Additionally, some genetic syndromes and chromosomal abnormalities may cause growth disorders.3
Depending on the molecular defect and clinical manifestations, IEI can impair growth through different mechanisms and, in some cases, several simultaneous mechanisms. This group of diseases should, therefore , be considered in the differential diagnosis of short stature and growth disorders.
Early diagnosis and treatment of IEI improve their prognosis; knowledge on the mechanisms through which growth can be impaired in this group of diseases allows specific treatment that improves growth of patients.
ObjectiveThis study aimed to review the literature on the repercussions of the different IEI on growth, drawing attention to the diagnosis of this group of diseases in patients with growth disorders, as well as to enable the identification of the different causes of growth disorders in patients with IEI.
MethodsA non-systematic review of the literature was carried out searching articles published in the last 18 years (since 2000) in PubMed with the terms “growth”, “growth disorders”, “failure to thrive”, or “short stature” AND “immunologic deficiency syndromes”, “immune deficiency disease”, or “immune deficiency” NOT HIV. The authors used filters to narrow the search to review articles in English or French. The Online Mendelian Inheritance in Man (OMIN) database was also searched for immune deficiencies and short stature or failure to thrive.
Weight-for-height growth is assessed by the weight, height/length, cephalic perimeter, and body mass index (BMI) measurements, included in charts of the World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC).4 Other measurements related to growth are body proportion, bone maturity, and dental development assessments.4 In this study, the authors analyzed the disorders associated with the IEI involving these measurements, except for BMI and cephalic perimeter.
ResultsGrowth is a complex process in which several genetic and environmental factor can play a role.5 Thus, an individual's growth depends on a sum of conditions in order to progress properly and completely. Among these factors, proper intake of nutrients, capacity to absorb these nutrients, inherited genetic potential, and integrity of the endocrine and osteoarticular pathways are noteworthy. Another critical aspect is the natural balance between energy sources and caloric expenditures.
Genetic disorders can affect hormonal function or osteoarticular system. Acquired disorders of growth are related to psychosocial factors and/or different diseases.3 Acquired causes of insufficient growth are related to endocrine disorders, low caloric intake, malabsorption, and increased caloric expenditure (such as infectious, inflammatory, or neoplastic processes).
Generally, in children with chronic diseases, growth failure is related to effects from poor nutrition and caloric expenditure resulting from the inflammatory process caused by the disease itself. Chronic malnutrition and release of inflammatory cytokines are determinant for GH-resistance.6 Proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), act on the central nervous system by changing the pathways of appetite and energy metabolism, causing muscle loss.7
The GH/insulin-like growth factor-1 (IGF1) axis plays a critical role in growth. Changes in metabolism and resistance of the organs to GH have been described as one of the main factors contributing to growth retardation in patients with inflammatory bowel disease in childhood (a common condition in patients with IEI).8 Various cytokines observed in inflammatory processes (of autoimmune or infectious nature) inhibit the pathways involving IGF-1. Increased IL6, generally present in chronic inflammatory conditions, appears to represent one of the main mechanisms affecting skeletal development.9
In primary immunodeficiencies, the vast majority of children present an increased number of infections and/or severe infections, requiring a persistent or recurring inflammatory response, stimulating a great number of cytokines. Accordingly, other IEIs present changes in the immune system regulation, which is associated with a reduction or absence of the control mechanisms from the immunological system itself, resulting in a chronic inflammatory process of variable intensity, according to the specific immune defect. Furthermore, the presence of inflammatory bowel disease in children with IEI also promotes reduced absorption of nutrients, which worsens the condition.
In addition to the relation of inflammatory process with growth and nutrition, several patients with IEI present genetic syndromes associated with short stature, such as chromosomal abnormalities, DNA repair defects, and osteoarticular dysplasia. Moreover, many IEI can be associated with endocrine system diseases that change the level of hormones essential to a child's normal growth, causing growth failure.
In short, the IEI can affect growth through different mechanisms and some of these immunity defects can change growth through multiple simultaneous mechanisms. These mechanisms can be divided as follows:
- -
genetic syndromes;
- -
osteoarticular system disorders;
- -
endocrine system disorders;
- -
reduced caloric intake;
- -
catabolic processes.
Early IEI diagnosis has relevant prognostic implications. The ten signs described by the Jeffrey Modell Foundation have been published worldwide and is based on experts’ opinions (Table 1). Despite being widely used, no studies have confirmed their efficacy on the clinical practice.10 Some studies have indicated that family history of immunodeficiency combined with use of venous antibiotics, deep infections, failure to thrive, early death of siblings, and consanguinity between parents were defined as best predictors of child IEI.10,11
Ten warning signs for IEI from the Jeffrey Modell Foundation, adapted to Brazil.
| Two or more pneumonias per year |
| Four or more otitis in one year |
| Recurrent stomatitis or moniliasis for more than two months |
| Recurrent abscesses or ecthyma |
| One deep systemic infection (meningitis, osteoarthritis, septicemia) |
| Recurrent intestinal infections/chronic diarrhea |
| Severe asthma, collagen disease, or autoimmune disease |
| Adverse effect to BCG and/or mycobacterial infection |
| Clinical phenotype suggestive of immunodeficiency syndrome |
| Family history of immunodeficiency |
Source: http://www.bragid.org.br/.
The initial investigation of IEI consists of complete blood count, serum immunoglobulins (A, M, G, and E) levels, lymphocyte subpopulations (CD3, CD4, CD8, CD19, and CD56/16) count, CH50 level (total hemolytic complement activity), and DHR (Dihydrorhodamine) test to evaluate neutrophil oxidative burst.11
The analysis of growth charts can help in the investigation of IEI and the mechanisms through which growth is affected. It is relevant to distinguish whether growth impairment is already present at birth, due to retarded intrauterine growth, or whether the patient is eutrophic in early childhood and presents impaired growth later, as it is relevant to observe the relationship between weight and length/height charts, in addition to body proportion.5,12
Growth disorders caused by genetic diseases (osteoarticular or chromosomal disorders) affect the charts since birth: the patient is born small and stays below the curves throughout childhood.5,12 Short stature with alterations in the proportion between trunk and limbs is, in general, associated with bone dysplasia.13
In endocrine disorders, height is affected before or simultaneously with weight, and the weight-for-height ratio is normal or increased. In nutritional defects (low ingestion, alteration in absorption, or catabolism), weight is affected before height and the weight-for-height ratio is low.3 In these disorders, delayed bone age is a usual finding.
The authors present below each one of the mechanisms involved in growth failure in patients with IEI, separately:
Genetic syndromes (with or without osteoarticular system disorders)Delayed intrauterine growth is commonly associated with IEI related to chromosomal disorders, bone dysplasia (bone formation disorders), and defects in DNA repair. In the latter, usually, patients also present microcephaly at birth.14
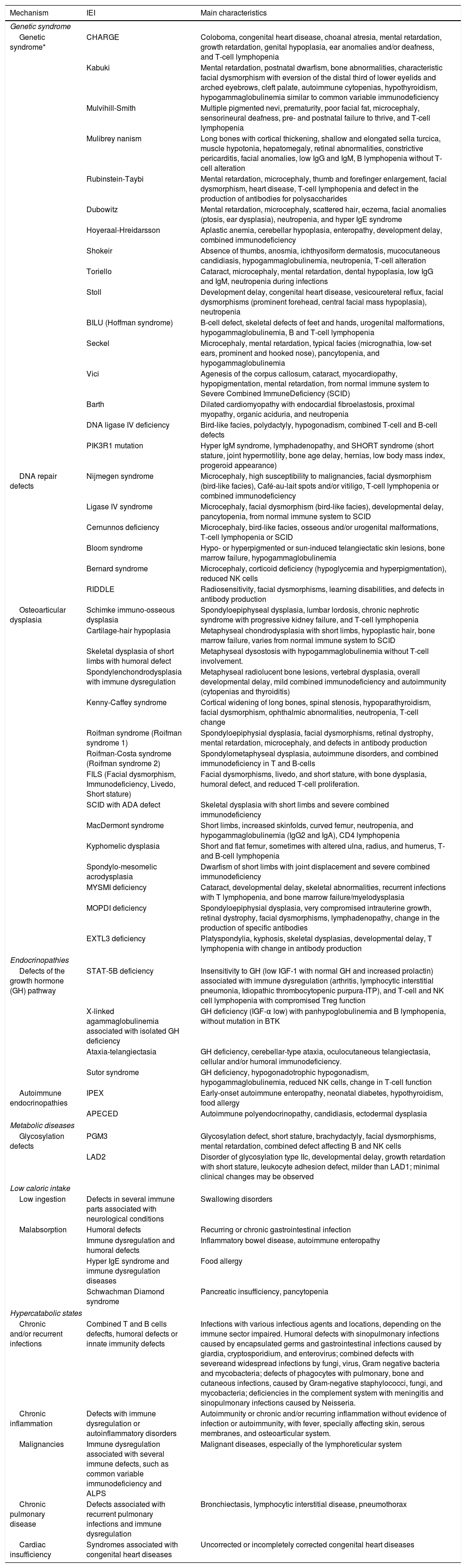
There are a large number of syndromes with defects of the immune system associated with short stature without changes in body proportion (Table 2).13 Several chromosomal diseases are associated with IEI, especially with defects in antibodies production.15
IEI and short stature: mechanisms, IEI, and main characteristics.
| Mechanism | IEI | Main characteristics |
|---|---|---|
| Genetic syndrome | ||
| Genetic syndrome* | CHARGE | Coloboma, congenital heart disease, choanal atresia, mental retardation, growth retardation, genital hypoplasia, ear anomalies and/or deafness, and T-cell lymphopenia |
| Kabuki | Mental retardation, postnatal dwarfism, bone abnormalities, characteristic facial dysmorphism with eversion of the distal third of lower eyelids and arched eyebrows, cleft palate, autoimmune cytopenias, hypothyroidism, hypogammaglobulinemia similar to common variable immunodeficiency | |
| Mulvihill-Smith | Multiple pigmented nevi, prematurity, poor facial fat, microcephaly, sensorineural deafness, pre- and postnatal failure to thrive, and T-cell lymphopenia | |
| Mulibrey nanism | Long bones with cortical thickening, shallow and elongated sella turcica, muscle hypotonia, hepatomegaly, retinal abnormalities, constrictive pericarditis, facial anomalies, low IgG and IgM, B lymphopenia without T-cell alteration | |
| Rubinstein-Taybi | Mental retardation, microcephaly, thumb and forefinger enlargement, facial dysmorphism, heart disease, T-cell lymphopenia and defect in the production of antibodies for polysaccharides | |
| Dubowitz | Mental retardation, microcephaly, scattered hair, eczema, facial anomalies (ptosis, ear dysplasia), neutropenia, and hyper IgE syndrome | |
| Hoyeraal-Hreidarsson | Aplastic anemia, cerebellar hypoplasia, enteropathy, development delay, combined immunodeficiency | |
| Shokeir | Absence of thumbs, anosmia, ichthyosiform dermatosis, mucocutaneous candidiasis, hypogammaglobulinemia, neutropenia, T-cell alteration | |
| Toriello | Cataract, microcephaly, mental retardation, dental hypoplasia, low IgG and IgM, neutropenia during infections | |
| Stoll | Development delay, congenital heart disease, vesicoureteral reflux, facial dysmorphisms (prominent forehead, central facial mass hypoplasia), neutropenia | |
| BILU (Hoffman syndrome) | B-cell defect, skeletal defects of feet and hands, urogenital malformations, hypogammaglobulinemia, B and T-cell lymphopenia | |
| Seckel | Microcephaly, mental retardation, typical facies (micrognathia, low-set ears, prominent and hooked nose), pancytopenia, and hypogammaglobulinemia | |
| Vici | Agenesis of the corpus callosum, cataract, myocardiopathy, hypopigmentation, mental retardation, from normal immune system to Severe Combined ImmuneDeficiency (SCID) | |
| Barth | Dilated cardiomyopathy with endocardial fibroelastosis, proximal myopathy, organic aciduria, and neutropenia | |
| DNA ligase IV deficiency | Bird-like facies, polydactyly, hypogonadism, combined T-cell and B-cell defects | |
| PIK3R1 mutation | Hyper IgM syndrome, lymphadenopathy, and SHORT syndrome (short stature, joint hypermotility, bone age delay, hernias, low body mass index, progeroid appearance) | |
| DNA repair defects | Nijmegen syndrome | Microcephaly, high susceptibility to malignancies, facial dysmorphism (bird-like facies), Café-au-lait spots and/or vitiligo, T-cell lymphopenia or combined immunodeficiency |
| Ligase IV syndrome | Microcephaly, facial dysmorphism (bird-like facies), developmental delay, pancytopenia, from normal immune system to SCID | |
| Cernunnos deficiency | Microcephaly, bird-like facies, osseous and/or urogenital malformations, T-cell lymphopenia or SCID | |
| Bloom syndrome | Hypo- or hyperpigmented or sun-induced telangiectatic skin lesions, bone marrow failure, hypogammaglobulinemia | |
| Bernard syndrome | Microcephaly, corticoid deficiency (hypoglycemia and hyperpigmentation), reduced NK cells | |
| RIDDLE | Radiosensitivity, facial dysmorphisms, learning disabilities, and defects in antibody production | |
| Osteoarticular dysplasia | Schimke immuno-osseous dysplasia | Spondyloepiphyseal dysplasia, lumbar lordosis, chronic nephrotic syndrome with progressive kidney failure, and T-cell lymphopenia |
| Cartilage-hair hypoplasia | Metaphyseal chondrodysplasia with short limbs, hypoplastic hair, bone marrow failure, varies from normal immune system to SCID | |
| Skeletal dysplasia of short limbs with humoral defect | Metaphyseal dysostosis with hypogammaglobulinemia without T-cell involvement. | |
| Spondylenchondrodysplasia with immune dysregulation | Metaphyseal radiolucent bone lesions, vertebral dysplasia, overall developmental delay, mild combined immunodeficiency and autoimmunity (cytopenias and thyroiditis) | |
| Kenny-Caffey syndrome | Cortical widening of long bones, spinal stenosis, hypoparathyroidism, facial dysmorphism, ophthalmic abnormalities, neutropenia, T-cell change | |
| Roifman syndrome (Roifman syndrome 1) | Spondyloepiphysial dysplasia, facial dysmorphisms, retinal dystrophy, mental retardation, microcephaly, and defects in antibody production | |
| Roifman-Costa syndrome (Roifman syndrome 2) | Spondylometaphyseal dysplasia, autoimmune disorders, and combined immunodeficiency in T and B-cells | |
| FILS (Facial dysmorphism, Immunodeficiency, Livedo, Short stature) | Facial dysmorphisms, livedo, and short stature, with bone dysplasia, humoral defect, and reduced T-cell proliferation. | |
| SCID with ADA defect | Skeletal dysplasia with short limbs and severe combined immunodeficiency | |
| MacDermont syndrome | Short limbs, increased skinfolds, curved femur, neutropenia, and hypogammaglobulinemia (IgG2 and IgA), CD4 lymphopenia | |
| Kyphomelic dysplasia | Short and flat femur, sometimes with altered ulna, radius, and humerus, T- and B-cell lymphopenia | |
| Spondylo-mesomelic acrodysplasia | Dwarfism of short limbs with joint displacement and severe combined immunodeficiency | |
| MYSMI deficiency | Cataract, developmental delay, skeletal abnormalities, recurrent infections with T lymphopenia, and bone marrow failure/myelodysplasia | |
| MOPDI deficiency | Spondyloepiphysial dysplasia, very compromised intrauterine growth, retinal dystrophy, facial dysmorphisms, lymphadenopathy, change in the production of specific antibodies | |
| EXTL3 deficiency | Platyspondylia, kyphosis, skeletal dysplasias, developmental delay, T lymphopenia with change in antibody production | |
| Endocrinopathies | ||
| Defects of the growth hormone (GH) pathway | STAT-5B deficiency | Insensitivity to GH (low IGF-1 with normal GH and increased prolactin) associated with immune dysregulation (arthritis, lymphocytic interstitial pneumonia, Idiopathic thrombocytopenic purpura-ITP), and T-cell and NK cell lymphopenia with compromised Treg function |
| X-linked agammaglobulinemia associated with isolated GH deficiency | GH deficiency (IGF-α low) with panhypoglobulinemia and B lymphopenia, without mutation in BTK | |
| Ataxia-telangiectasia | GH deficiency, cerebellar-type ataxia, oculocutaneous telangiectasia, cellular and/or humoral immunodeficiency. | |
| Sutor syndrome | GH deficiency, hypogonadotrophic hypogonadism, hypogammaglobulinemia, reduced NK cells, change in T-cell function | |
| Autoimmune endocrinopathies | IPEX | Early-onset autoimmune enteropathy, neonatal diabetes, hypothyroidism, food allergy |
| APECED | Autoimmune polyendocrinopathy, candidiasis, ectodermal dysplasia | |
| Metabolic diseases | ||
| Glycosylation defects | PGM3 | Glycosylation defect, short stature, brachydactyly, facial dysmorphisms, mental retardation, combined defect affecting B and NK cells |
| LAD2 | Disorder of glycosylation type IIc, developmental delay, growth retardation with short stature, leukocyte adhesion defect, milder than LAD1; minimal clinical changes may be observed | |
| Low caloric intake | ||
| Low ingestion | Defects in several immune parts associated with neurological conditions | Swallowing disorders |
| Malabsorption | Humoral defects | Recurring or chronic gastrointestinal infection |
| Immune dysregulation and humoral defects | Inflammatory bowel disease, autoimmune enteropathy | |
| Hyper IgE syndrome and immune dysregulation diseases | Food allergy | |
| Schwachman Diamond syndrome | Pancreatic insufficiency, pancytopenia | |
| Hypercatabolic states | ||
| Chronic and/or recurrent infections | Combined T and B cells defecfts, humoral defects or innate immunity defects | Infections with various infectious agents and locations, depending on the immune sector impaired. Humoral defects with sinopulmonary infections caused by encapsulated germs and gastrointestinal infections caused by giardia, cryptosporidium, and enterovirus; combined defects with severeand widespread infections by fungi, virus, Gram negative bacteria and mycobacteria; defects of phagocytes with pulmonary, bone and cutaneous infections, caused by Gram-negative staphylococci, fungi, and mycobacteria; deficiencies in the complement system with meningitis and sinopulmonary infections caused by Neisseria. |
| Chronic inflammation | Defects with immune dysregulation or autoinflammatory disorders | Autoimmunity or chronic and/or recurring inflammation without evidence of infection or autoimmunity, with fever, specially affecting skin, serous membranes, and osteoarticular system. |
| Malignancies | Immune dysregulation associated with several immune defects, such as common variable immunodeficiency and ALPS | Malignant diseases, especially of the lymphoreticular system |
| Chronic pulmonary disease | Defects associated with recurrent pulmonary infections and immune dysregulation | Bronchiectasis, lymphocytic interstitial disease, pneumothorax |
| Cardiac insufficiency | Syndromes associated with congenital heart diseases | Uncorrected or incompletely corrected congenital heart diseases |
Most genetic syndromes, with or without osteoarticular involvement, are listed in Tables 1 and 2 of the IEI classification, which include combined defects of T- and B-cells and combined defects associated with the syndromes, respectively.1 Genetic defects in proteins involved in DNA repair are usually associated with immunological abnormalities, which range from a severe impairment, with a phenotype of severe combined immunodeficiency (as is the case of ligase IV deficiency and Cernunnos deficiency) to milder defects. Defects in GINS complex, essential for DNA replication prior to cell division, particularly affect neutrophils and NK cells, producing a phenotype different from the combined immunodeficiency.16–18
Osteoarticular system disordersBone dysplasias affect bone and growth cartilage; they present specific radiological findings depending on the genetic defect (Table 2) and can produce, in addition to impaired growth, changes in body proportion and deformities.13
Dysplasias associated with immune system disorders are referred to as immuno-osseous dysplasias and are related to varying levels of T- and/or B-cell deficiency. There are reports of hypochondroplasia (less severe skeletal changes than in achondroplasia) and other immunological defects, such as CD4 lymphopenia and IgA deficiency.19
Patients with cartilage-hair hypoplasia show severe short stature, short limbs, ectodermal dysplasia, anemia, variable immunodeficiency (generally combined, later onset), and increased susceptibility to malignancies.20 The radiological findings are quite variable, but, characteristically, they have short and wide bones, with prominent and irregular metaphyses and globular epiphyses on knees and ankles.21
Other immuno-osseous dysplasias are short-limb skeletal dysplasia with combined immunodeficiency, MacDermot syndrome, kyphomelic dysplasia, spondyl-mesomelic acrodysplasia, short-limb skeletal dysplasia with humoral immunodeficiency, Schimke dysplasia, Roifman syndrome, SPENCDI syndrome, Kenny-Caffey syndrome, MYSMI deficiency, MOPDI deficiency, and EXTL3 deficiency1,13 (Table 2).
In combined immunodeficiency due to ADA deficiency, there are reports of short stature with short limbs and costal deformities that can be at least partially reversed with enzyme replacement therapy, bone marrow transplantation, or gene therapy.22
Infectious conditions (more common in phagocyte defects) or inflammatory (autoinflammatory diseases) can also cause asymmetric limb growth, with changes in body proportions and/or localized deformities.23
Endocrine system disordersEndocrine disorders related to IEI can be autoimmune in nature or related to changes in GH pathway. The latter includes the STAT5b deficiency and agammaglobulinemia with GH deficiency.
IEI characterized by autoimmunity or immune dysregulation cause impact on growth due to secondary endocrinopathies. Most autoimmune endocrine diseases, particularly thyroid and parathyroid diseases and diabetes mellitus, are listed on Table 4 of the IEI classification, which addresses immune dysregulation.1
In STAT5b defect, there is impaired insulin-like growth factor 1 (IGF1) production, which is phenotypically similar to GH insensitivity syndrome (Laron syndrome).24 More recently, dominant monoallelic mutations have been described.24,25 There is a reduction in the dosages of IGF1, IGF-binding protein-3 (IGFBP3) and acid-labile subunit (ALS), and a significant increase in prolactin (IGFBP3 acid labile subunit).24,26 This deficiency is also associated with eczema, chronic lung disease (lymphocytic pneumonia, fibrosis), hypergammaglobulinemia, and T-cell and Treg lymphopenia.27
Patients with mutations with gain of function of STAT3 can also present short stature; the mechanisms through which it occurs are not fully understood.28 It is possible that it occurs through STAT5 activation and partial insensitivity to GH.29,30 These patients, however, present multiple early manifestations of autoimmunity, requiring immunosuppressive treatment, which hinders differentiation from short stature due to the chronic/recurrent use of systemic corticosteroids.
Other defects in cytokine signaling that can manifest with short stature are those of the PI3K pathway.31,32 The changed signaling in the PI3K-AKT-mTOR pathway may lead to insulin- and growth factor-resistance, with impaired cell division and consequent growth retardation.33,34 Patients with PI3KR1 mutation may present SHORT (short stature, joint hyperextensibility, teething delay, partial lipodystrophy) syndrome, as well as hyper IgM syndrome, and lymphadenopathy.29,30
Agammaglobulinemia and X-linked isolated GH deficiency presents many similarities to X-linked agamagobulinemia, with panhypoglobulinemia and low levels of B lymphocytes, but there is no mutation or altered expression of BTK.35,36
Reduction in central GH secretion has also been described in ataxia-telangiectasia.37
Reduced caloric intakeChanges in caloric intake may be due to simple poor nutrient ingestion or swallowing disorders in IEI that evolve with motor neurological disorders, such as ataxia-telangiectasia.1
In other IEI, there may be significant malabsorption of nutrients related to an inflammatory process of the digestive tract (inflammatory disease), of infectious or allergic nature, or even caused by pancreatic dysfunction.1
Catabolic processesTo a greater or lesser extent, catabolic processes are involved in diseases of virtually all Tables of the IEI classification.
Acute weight changes are particularly related to acute infectious and/or inflammatory conditions and malignancies. Usually, patients are able to reach the normal curves once the process is controlled.
However, chronic inflammatory/infectious conditions, as well as gastrointestinal losses, cause more persistent impairment in growth curves. Malignancies, chronic lung disease, and heart failure can contribute to the onset of a hypercatabolic condition.
In most IEI, more than one factor contributes to growth impairment. An example would be IPEX syndrome, in which endocrine diseases (autoimmune hypothyroidism, diabetes mellitus), loss of nutrients (autoimmune enteropathy), and low nutrient ingestion (food allergy) contribute to growth impairment.
Table 2 presents a summary of the mechanisms involved in growth failure in several IEI and their main characteristics.
Specific changes in dental development can be observed in some IEI. The following are the most commonly described alterations: dental hypoplasia in ectodermal dysplasia with immunodeficiency (NF-kB essential modular- NEMO and others), delayed tooth replacement in autosomal dominant hyper IgE syndrome, and early tooth decay in cyclic neutropenia.1
Changes in bone maturity are well described in thyroid and/or parathyroid endocrinopathies, mainly associated with IEI with immune dysregulation. Chronic diseases of any type, including infectious/inflammatory conditions typical of IEI, usually promote delayed bone maturation assessed by bone age.38 Changes in IGF1 production are observed in cases of malnutrition, inflammatory bowel disease, and liver diseases,5 which can be part of the clinical condition of many IEI.
Once the diagnosis of IEI has been confirmed, adequate nutritional intake, including assessment of the need for individualized supplementation for each patient, control of infections and inflammatory process, and monitoring and treatment of those patients with syndromes associated with endocrine disorders, are important in order to keep patients' growth as good as possible. Patients with syndromic disorders associated with the osteoarticular system disease should be early identified, in order to initiate orthopedic-physiotherapeutic measures to minimize the impact of such malformations.39
ConclusionIn general, patients with IEI have a higher risk of growth failure. The type of IEI allows us to anticipate what type of growth disorder we can expect. In turn, the type of growth disorder can help in the diagnosis of clinical conditions related to IEI. In many IEI, however, the causes of poor growth are mixed, involving more than one factor.
In patients who are below the growthcharts since birth, genetic syndromes associated with defects in the immune system should be considered, with or without osteoarticular disorders.
Postnatal changes in growth, in which early height impairment is observed, should lead physicians to consider endocrine disorders associated with IEI or osteoarticular diseases. In the latter, it is important to be alert for changes in body proportion or deformities (Fig. 1).

Even nowadays, the lack of early recognition of IEI leads to a late diagnosis, depriving patients of early appropriate treatment, with undesirable consequences to the growth of children and adolescents. It is essential to be alert to the warning signs for IEI in face of growth disorders (Table 3).
Warning signs for IEI in face of growth disorders.
| Family history of IEI |
| Recurring, severe infections and/or hardly responsive to treatment and/or caused by opportunistic germs |
| Use of venous antibiotics for sepsis |
| Two or more autoimmune endocrinopathies |
| Early autoimmune endocrinopathy |
| Multiple and/or early autoimmunity manifestations |
| Recurring and/or persistent fever |
| Severe allergy of difficult control |
| Persistent diarrhea, with or without infection |
| Clinical phenotype suggestive of syndromes associated with IEI |
| Early death of sibling |
| Hypogammaglobulinemia in protein electrophoresis |
| Lymphopenia in blood count |
Proper and early diagnosis and treatment with a multidisciplinary team (including a nutritionist and physical therapist) are important to maintain, as much as possible, adequate patient growth. Furthermore, in many cases, impaired growth can be adjusted through adequate IEI treatment.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Goudouris ES, Segundo GR, Poli C. Repercussions of inborn errors of immunity on growth. J Pediatr. 2019;95:S49–S58.




