


To identify chromosomal imbalances by whole‐genome microarray‐based comparative genomic hybridization (array‐CGH) in DNA samples of neonates with congenital anomalies of unknown cause from a birth defects monitoring program at a public maternity hospital.
MethodsA blind genomic analysis was performed retrospectively in 35 stored DNA samples of neonates born between July of 2011 and December of 2012. All potential DNA copy number variations detected (CNVs) were matched with those reported in public genomic databases, and their clinical significance was evaluated.
ResultsOut of a total of 35 samples tested, 13 genomic imbalances were detected in 12/35 cases (34.3%). In 4/35 cases (11.4%), chromosomal imbalances could be defined as pathogenic; in 5/35 (14.3%) cases, DNA CNVs of uncertain clinical significance were identified; and in 4/35 cases (11.4%), normal variants were detected. Among the four cases with results considered causally related to the clinical findings, two of the four (50%) showed causative alterations already associated with well‐defined microdeletion syndromes. In two of the four samples (50%), the chromosomal imbalances found, although predicted as pathogenic, had not been previously associated with recognized clinical entities.
ConclusionsArray‐CGH analysis allowed for a higher rate of detection of chromosomal anomalies, and this determination is especially valuable in neonates with congenital anomalies of unknown etiology, or in cases in which karyotype results cannot be obtained. Moreover, although the interpretation of the results must be refined, this method is a robust and precise tool that can be used in the first‐line investigation of congenital anomalies, and should be considered for prospective/retrospective analyses of DNA samples by birth defect monitoring programs.
Identificar desequilíbrios cromossômicos por meio da hibridização genômica comparativa baseada em microarranjos (CGH‐array) em amostras de DNA de neonatos com anomalias congênitas de causa desconhecida de um programa de monitoramento de defeitos congênitos em uma maternidade pública.
MétodosUma análise genômica cega foi realizada retrospectivamente em 35 amostras armazenadas de DNA de neonatos nascidos entre julho de 2011 e dezembro de 2012. Todas as possíveis variações no número de cópias (CNVs) de DNA foram comparadas com as relatadas em bases de dados genômicos públicas, e sua relevância clínica foi avaliada.
ResultadosDe um total de 35 amostras testadas, foram detectados 13 desequilíbrios genômicos em 12/35 casos (34,3%). Em 4/35 casos (11,4%), os desequilíbrios cromossômicos poderiam ser definidos como patogênicos; em 5/35 (14,3%) deles foram identificadas CNVs de DNA de relevância clínica incerta; e, em 4/35 (11,4%), foram detectadas variações normais. Dentre os quatro casos com resultados considerados relacionados causalmente aos achados clínicos, 2/4 (50%) apresentaram alterações causais já relacionadas a síndromes de microdeleção bem definidas. Em 2/4 amostras (50%), os desequilíbrios cromossômicos encontrados, embora preditivos como patogênicos, não estavam relacionados anteriormente a entidades clínicas reconhecidas.
ConclusõesA análise de CGH‐array permitiu maior taxa de detecção de anomalias cromossômicas, e essa determinação é valiosa principalmente em neonatos com anomalias congênitas de etiologia desconhecida ou em casos em que os resultados do cariótipo não podem ser obtidos. Além disso, embora a interpretação dos resultados deva ser refinada, esse método é uma ferramenta robusta e precisa que pode ser usada na investigação de primeira linha de anomalias congênitas e deve ser considerada em análises futuras/retrospectivas de amostras de DNA por programas de monitoramento de defeitos congênitos.
Embora causas mendelianas, cromossômicas e ambientais tenham sido estabelecidas para muitas anomalias congênitas e transtornos dismórficos corporais, a etiologia precisa de várias dessas doenças ainda não foi identificada. Investigações etiológicas de anomalias congênitas sugerem que 6% dos defeitos congênitos estão relacionados a anomalias cromossômicas.1 No entanto, a proporção de anomalias cromossômicas em defeitos congênitos pode ser maior. Alguns indivíduos com anomalias congênitas podem ter desequilíbrios genômicos abaixo da resolução (> 5 Mb) da análise cromossômica padrão. Na última década, ocorreram desenvolvimentos significativos na detecção molecular de desequilíbrios cromossômicos, e a sua relação casual com anomalias congênitas e deficiências mentais aumentou. A lacuna considerável entre a resolução para detecção de anomalias cromossômicas com microscopia óptica e a análise genética molecular foram preenchidas com a introdução de abordagens moleculares, tais como a hibridização genômica comparativa em microarranjos (CGH‐array). Atualmente, o CGH‐Array é um método poderoso para a detecção simultânea de desequilíbrios cromossômicos e anomalias cromossômicas mais frequentes. O CGH‐Array permite a detecção de trissomias e grandes anomalias cromossômicas (já reconhecidas por análise padrão de cariótipo), bem como de desequilíbrios cromossômicos submicroscópicos (deleções, duplicações ou triplicações de qualquer região cromossômica, algumas das quais são reconhecidas por hibridização in situ por fluorescência (FISH)) que resultam em variações no número de cópias (CNVs). Vários estudos mostraram que o uso de tecnologias baseadas em array aumentou a taxa de detecção de anomalias cromossômicas para, aproximadamente, 14%‐18% em comparação a uma taxa de aproximadamente 3% (excluindo a trissomia 21) usando abordagens citogenéticas padrão em indivíduos com atrasos no desenvolvimento, deficiências intelectuais, dificuldades de aprendizagem, anomalias congênitas múltiplas (ACMs), transtornos do espectro autístico, esquizofrenia e outras doenças neuropsiquiátricas.2 A frequência geral de anomalias cromossômicas não balanceadas foi relatada como 0,43% em neonatos, de acordo com relatos recentes.3,4 Portanto, a introdução da análise de todo o genoma por CGH‐array no período neonatal, quando alguns achados clínicos referentes a causas reconhecidas podem estar presentes, possivelmente aumenta a possibilidade de detecção precoce de anomalias cromossômicas compatíveis com uma doença genética/genômica.
Portanto, o objetivo deste estudo foi identificar desequilíbrios cromossômicos por meio de uma análise retrospectiva de todo o genoma usando hibridização genômica comparativa em microarranjos (CGH‐array) nas amostras de DNA armazenadas de neonatos com anomalias congênitas de causa desconhecida. Além disso, este estudo avaliou a contribuição do CGH‐array como uma ferramenta de diagnóstico de primeira linha em neonatos com anomalias congênitas avaliados por um programa de monitoramento de defeitos congênitos em uma maternidade pública no Sul do Brasil.
MétodosSeleção de amostrasEste estudo (com uma abordagem retrospectiva) foi realizado usando amostras de DNA anonimizadas extraídas do sangue de neonatos e obtidas do biorrepositório do Programa de Monitoramento de Defeitos Congênitos (PMDC) do Hospital de Clínicas de Porto Alegre (HCPA), Brasil. Os indivíduos tinham menos de 30 dias de vida e apresentavam uma ampla variedade de anomalias congênitas de causa desconhecida, com suspeita de anomalia cromossômica. As indicações clínicas para análise citogenética no momento de indicação foram retiradas de dados clínicos e laboratoriais coletados no nascimento e disponíveis em registros hospitalares e não incluíam investigações de acompanhamento ou informações sobre a evolução da doença. Casos com dados clínicos insuficientes foram excluídos, como aqueles em que as mães tinham suspeita clínica ou laboratorial de doenças infecciosas/parasitárias ou um histórico de uso/abuso de drogas ilícitas/álcool durante a gravidez. De acordo com esses critérios, um total de 45 amostras foi selecionado, porém dez destas foram excluídas por não possuírem a qualidade ideal de DNA necessária para a análise de CGH‐array, sendo o estudo realizado com 35 amostras. Os resultados das análises cromossômicas anteriores foram obtidos em 32 casos. O teste citogenético convencional na resolução de 500‐550 bandas foi inicialmente normal em todos os casos, porém, em um deles, um relatório de um cariótipo anormal foi fornecido posteriormente. Este estudo foi aprovado pelo Conselho de Revisão Institucional do HCPA e conduzido de acordo com normas éticas institucionais vigentes a respeito do uso de materiais biológicos de biorrepositórios.5
CGH‐Array de todo o genomaRealizamos um CGH‐Array de oligonucleotídeos usando uma plataforma 8x60K em todo o genoma (modelo 021924, Agilent Technologies, Santa Clara, EUA), com uma média de espaçamento de 40 Kb entre as sondas. O DNA genômico foi isolado do sangue periférico de 35 neonatos, fornecido pelo PMDC‐HCPA, e analisado posteriormente. Para cada experimento, foi usada uma referência normal de sexo diferente (Promega Corp Madison, WI, EUA). Os experimentos foram realizados de acordo com o protocolo do fabricante. As imagens dos arrays foram obtidas usando‐se um scanner de microarrays (modelo G2600D, Agilent, Califórnia, EUA) e processadas por meio do software Feature Extraction (modelo v9.5.1, Agilent, Califórnia, EUA). Os dados brutos foram analisados pelo software Agilent Cytogenomics (modelo v2.7.8.0, Agilent, Califórnia, EUA) com o algoritmo estatístico ADM‐2, usando um limite de 6,0 e uma ocorrência de anomalia mínima em quatro sondas. Foi realizada a normalização subsequente dos dados por software para verificação de variações no número de cópias de DNA. Os valores de p para cada sonda foram calculados, fornecendo critérios estatísticos objetivos adicionais para determinar se o desvio de cada sonda de zero era estatisticamente significativo.6 Todas as experiências incluíram duas hibridizações em array por amostra, e os resultados foram registrados e comparados. Foram relatados apenas desequilíbrios genômicos detectados em ambas as experiências de inversão de corantes (dye‐swap).
Análise de dadosAs análises dos dados do CGH‐array de todo o genoma foram realizadas de maneira cega; as amostras foram recebidas e anonimizadas, e os investigadores que realizaram as análises de CGH‐array não tinham conhecimento das informações clínicas e laboratoriais anteriores relacionadas a cada amostra. As variações no número de cópias (CNVs) de DNA detectadas foram comparadas com as CNVs relatadas em recursos on‐line disponíveis publicamente e bases de dados de anomalias e variações cromossômicas.7–13 Classificamos as CNVs (ganho/duplicação ou perdas/deleções) em diferentes categorias: CNV benigna (variação genômica normal); CNV de relevância clínica incerta (variação de relevância incerta (VOUS)); e CNV de possível relevância clínica (variação patogênica). Neste estudo, as anomalias patogênicas incluíam a detecção de CNVs em regiões patogênicas conhecidas, deleção/duplicação > 3 Mb em tamanho ou visível por meio de um cariótipo com bandeamento G não relatado na população normal e deleções ou duplicações < 3 Mb anteriormente relatadas como patogênicas. As deleções ou duplicações benignas incluíam as variações bem documentadas na população normal ou anteriormente relatadas como benignas. As deleções ou duplicações foram classificadas como VOUS quando evidências insuficientes estavam disponíveis para concluir se a CNV era patogênica ou benigna.
ResultadosOs dados de 35 neonatos com anomalias congênitas de causa desconhecida, nascidos entre julho de 2011 e dezembro de 2012, cujas amostras de DNA foram analisadas por meio do CGH‐array de todo o genoma, são apresentados na tabela 1. A idade materna variou de 19 a 41 anos de idade. Identificamos 12 (34,3%) casos com variações no número de cópias (CNVs) de DNA. Desses casos, 7/12 (58,3%) eram do sexo masculino e 5/12 (41,7%) do sexo feminino. Os detalhes dos resultados do CGH‐array dos casos com desequilíbrios genômicos estão resumidos na tabela 2. Foram identificadas 13 CNVs em 12 indivíduos. Em geral, foram verificadas duplicações em 6/35 (17%) e deleções em 7/35 (20%) dos casos. Em 6/35 casos (17%), foi identificada apenas uma deleção; 5/35 casos (14,3%) apresentaram apenas uma duplicação, e 1/35 (2,8%) apresentou uma deleção e uma duplicação. Além disso, um teste de hibridização in situ por fluorescência (FISH) confirmou os resultados do CGH‐array em um caso de deleção (caso 14), cujas células armazenadas ficaram disponíveis (dados não apresentados).
Resumo das indicações clínicas das 35 amostras no momento de indicação para análise cromossômica
| Caso n° | Principais características clínicas relacionadas |
|---|---|
| 1a | Sexo feminino, hérnia diafragmática congênita (HDC), microtia, hipertelorismo, cardiopatia congênita (CC) (dextroposição do coração), atresia anal |
| 2 | Sexo masculino, CC (Tetralogia de Fallot) |
| 4 | Sexo masculino, onfalocele, microcefalia |
| 5 | Sexo masculino, onfalocele, agenesia de membros, genitália ambígua, atresia anal, disfunção da bexiga |
| 10 | Sexo feminino, gastrosquise |
| 12a | Sexo masculino, HDC |
| 13 | Sexo masculino, focomelia unilateral, displasia da anca |
| 14 | Sexo masculino, assimetria craniana, fissura palatina (mole), hipertelorismo ocular, atresia esofágica tipo IIIB, camptodactilia do 3°, 4° e 5° dedos, clinodactilia do 5° dedo, pés tortos |
| 15 | Sexo masculino, atresia anal, pés tortos |
| 16 | Sexo feminino, atresia anal, genitália hipoplásica, CC |
| 17 | Sexo feminino, artrogripose múltipla congênita (amioplasia) |
| 19 | Sexo feminino, fissura labial unilateral (esquerdo) e fissura palatina não sindrômicas |
| 22a | Sexo feminino, microcefalia, hipoplasia cerebelar, oligoidrâmnio, hipoplasia pulmonar, displasia renal, hipoplasia genital, microgiria frontal, encefalocele occipital |
| 23 | Sexo feminino, onfalocele, microcefalia |
| 24 | Sexo masculino, micrognatismo, incisivo central superior único |
| 25 | Sexo feminino, fissura labiopalatina (bilateral) não sindrômica |
| 29 | Sexo masculino, CC (Tetralogia de Fallot) |
| 30 | Sexo masculino, fissura labiopalatina (bilateral) não sindrômica |
| 31a | Sexo masculino, artrogripose múltipla congênita (amioplasia), CC (Tetralogia de Fallot), displasia da anca |
| 32a | Sexo feminino, rim displásico multicístico bilateral, oligoidrâmnio |
| 33 | Sexo masculino, fissura palatina, pés tortos |
| 34 | Sexo masculino, fissura labial (esquerdo) e fissura palatina, bico de viúva, mamilos muito espaçados, hipoplasia genital, hipospadia |
| 35 | Sexo feminino, holoprosencefalia (HPE), oligoidrâmnio, microcefalia, atresia coanal unilateral |
| 37 | Sexo masculino, fissura labiopalatina (bilateral) não sindrômica |
| 38 | Sexo masculino, atresia esofágica tipo IIIB |
| 40 | Sexo masculino, retardo do crescimento intrauterino |
| 41 | Sexo feminino, gastrosquise |
| 42 | Sexo feminino, CC (Tetralogia de Fallot) |
| 43 | Sexo masculino, fissura palatina não sindrômica |
| 44 | Sexo masculino, microgiria, lisencefalia incompleta, micrognatismo |
| 46 | Sexo masculino, meningocele, pé torto (esquerdo) |
| 47 | Sexo feminino, gastrosquise |
| 48a | Sexo masculino, rim displásico multicístico bilateral, oligoidrâmnio, hipoplasia pulmonar bilateral |
| 49 | Sexo feminino, gastrosquise |
| 50 | Sexo masculino, artrogripose múltipla congênita, micrognatismo |
HDC, hérnia diafragmática congênita; CC, cardiopatia congênita; HPE, holoprosencefalia.
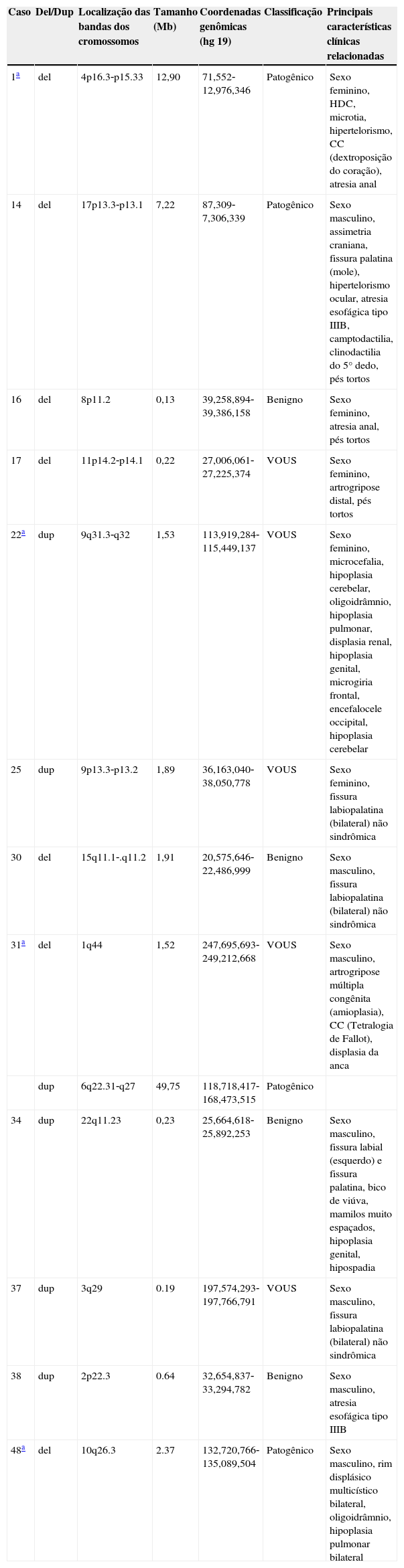
Detalhes do CGH‐array de 12 amostras com desequilíbrios cromossômicos
| Caso | Del/Dup | Localização das bandas dos cromossomos | Tamanho (Mb) | Coordenadas genômicas (hg 19) | Classificação | Principais características clínicas relacionadas |
|---|---|---|---|---|---|---|
| 1a | del | 4p16.3‐p15.33 | 12,90 | 71,552‐12,976,346 | Patogênico | Sexo feminino, HDC, microtia, hipertelorismo, CC (dextroposição do coração), atresia anal |
| 14 | del | 17p13.3‐p13.1 | 7,22 | 87,309‐7,306,339 | Patogênico | Sexo masculino, assimetria craniana, fissura palatina (mole), hipertelorismo ocular, atresia esofágica tipo IIIB, camptodactilia, clinodactilia do 5° dedo, pés tortos |
| 16 | del | 8p11.2 | 0,13 | 39,258,894‐39,386,158 | Benigno | Sexo feminino, atresia anal, pés tortos |
| 17 | del | 11p14.2‐p14.1 | 0,22 | 27,006,061‐27,225,374 | VOUS | Sexo feminino, artrogripose distal, pés tortos |
| 22a | dup | 9q31.3‐q32 | 1,53 | 113,919,284‐115,449,137 | VOUS | Sexo feminino, microcefalia, hipoplasia cerebelar, oligoidrâmnio, hipoplasia pulmonar, displasia renal, hipoplasia genital, microgiria frontal, encefalocele occipital, hipoplasia cerebelar |
| 25 | dup | 9p13.3‐p13.2 | 1,89 | 36,163,040‐38,050,778 | VOUS | Sexo feminino, fissura labiopalatina (bilateral) não sindrômica |
| 30 | del | 15q11.1‐.q11.2 | 1,91 | 20,575,646‐22,486,999 | Benigno | Sexo masculino, fissura labiopalatina (bilateral) não sindrômica |
| 31a | del | 1q44 | 1,52 | 247,695,693‐249,212,668 | VOUS | Sexo masculino, artrogripose múltipla congênita (amioplasia), CC (Tetralogia de Fallot), displasia da anca |
| dup | 6q22.31‐q27 | 49,75 | 118,718,417‐168,473,515 | Patogênico | ||
| 34 | dup | 22q11.23 | 0,23 | 25,664,618‐25,892,253 | Benigno | Sexo masculino, fissura labial (esquerdo) e fissura palatina, bico de viúva, mamilos muito espaçados, hipoplasia genital, hipospadia |
| 37 | dup | 3q29 | 0.19 | 197,574,293‐197,766,791 | VOUS | Sexo masculino, fissura labiopalatina (bilateral) não sindrômica |
| 38 | dup | 2p22.3 | 0.64 | 32,654,837‐33,294,782 | Benigno | Sexo masculino, atresia esofágica tipo IIIB |
| 48a | del | 10q26.3 | 2.37 | 132,720,766‐135,089,504 | Patogênico | Sexo masculino, rim displásico multicístico bilateral, oligoidrâmnio, hipoplasia pulmonar bilateral |
HDC, hérnia diafragmática congênita; CC, cardiopatia congênita; VOUS, variação de relevância incerta; CGH‐array, hibridização genômica comparativa em microarranjos; del, deleção; dup, duplicação.
Dentre os cinco indivíduos com fissuras orofaciais sindrômicas ou não sindrômicas (casos 14, 25, 30, 34 e 37) nos quais foram detectados desequilíbrios genômicos, um caso exibiu uma deleção clinicamente significativa de 7,2 Mb no cromossomo na região 17p13.3‐p13.1 (caso 14), que coincide com a região conhecida da síndrome de Miller‐Dieker (SMD). A análise FISH confirmou a deleção do cromossomo na região 17p13.3 (dados não apresentados). Os outros quatro casos com fissuras orofaciais apresentaram CNVs classificadas como benignas ou VOUS. Dos dois casos com artrogripose múltipla congênita (17 e 31), constatamos em um indivíduo uma duplicação intersticial do braço longo do cromossomo 6 na banda q22.31‐q27 e uma deleção terminal do braço longo do cromossomo 1 na banda q44. A análise de cariótipo anterior mostrou a identificação de uma anomalia cromossômica de origem desconhecida envolvendo o braço longo do cromossomo 6, porém não o desequilíbrio cromossômico que envolveu o cromossomo 1. Essa criança morreu com 35 dias de idade. Dos cinco casos adicionais com ACMs (1, 16, 22, 38 e 48), identificamos desequilíbrios cromossômicos clinicamente significativos ou possíveis CNVs patogênicas em três (1, 31 e 48). Os indivíduos morreram com as idades de dois dias, 5 horas e três dias após o nascimento, respectivamente. Em geral, as deleções foram classificadas como patogênicas em três casos (1, 14 e 48), como benignas em dois casos (16 e 30) e como VOUS em dois casos (17 e 31). As duplicações foram classificadas como patogênicas em um caso (31), como benignas em dois casos (34 e 38) e como VOUS em três casos (casos 22, 25 e 37). Os exemplos de visão geral gráfica do CGH‐array são apresentados na figura 1.
 e DNA de indivíduos normais como referência (em azul). São apresentados os dados das razões de teste/referência de cada cromossomo. Cada ponto representa uma única sonda (oligo) encontrada no array. A razão de log das sondas cromossômicas está traçada como uma função da posição cromossômica. A perda de número de cópias move a razão para a esquerda (valor de cerca de ‐1x). O ganho de número de cópias move a razão para a direita (valor de cerca de+1x). O ideograma de cada cromossomo (margem esquerda) mostra a localização de cada sonda. As razões de log2da sonda foram traçadas de acordo com as coordenadas genômicas (com base no UCSC Genome Browser, fevereiro de 2009, sequência de referência NCBI Build 37). A: A ∼ deleção terminal de 1,5 Mb no cromossomo 1q44 (linha azul) no caso 31. B: A ∼ deleção terminal de 12,9 Mb no cromossomo 4p16.3‐p15.33 (caixa azul) no caso 1. C: A ∼ duplicação intersticial de 49,7 Mb no cromossomo 6q22.31‐q37 (caixa azul) no caso 31. D: A ∼ deleção terminal de 2,37 Mb no cromossomo 10q26.3 (caixa azul) no caso 48. E: A ∼ deleção terminal de 7,2 Mb no cromossomo 17p13.3‐p13.1 (caixa azul) no caso 14.")
Perfis de taxa de CGH‐array dos cromossomos em quatro neonatos com desequilíbrios cromossômicos patogênicos usando DNA genômico dos neonatos como teste (em vermelho) e DNA de indivíduos normais como referência (em azul). São apresentados os dados das razões de teste/referência de cada cromossomo. Cada ponto representa uma única sonda (oligo) encontrada no array. A razão de log das sondas cromossômicas está traçada como uma função da posição cromossômica. A perda de número de cópias move a razão para a esquerda (valor de cerca de ‐1x). O ganho de número de cópias move a razão para a direita (valor de cerca de+1x). O ideograma de cada cromossomo (margem esquerda) mostra a localização de cada sonda. As razões de log2da sonda foram traçadas de acordo com as coordenadas genômicas (com base no UCSC Genome Browser, fevereiro de 2009, sequência de referência NCBI Build 37). A: A ∼ deleção terminal de 1,5 Mb no cromossomo 1q44 (linha azul) no caso 31. B: A ∼ deleção terminal de 12,9 Mb no cromossomo 4p16.3‐p15.33 (caixa azul) no caso 1. C: A ∼ duplicação intersticial de 49,7 Mb no cromossomo 6q22.31‐q37 (caixa azul) no caso 31. D: A ∼ deleção terminal de 2,37 Mb no cromossomo 10q26.3 (caixa azul) no caso 48. E: A ∼ deleção terminal de 7,2 Mb no cromossomo 17p13.3‐p13.1 (caixa azul) no caso 14.
O objetivo deste estudo foi identificar, retrospectivamente, desequilíbrios genômicos por meio de uma hibridização genômica comparativa em microarranjos (CGH‐array) de todo o genoma nas amostras disponíveis de neonatos com anomalias congênitas de causa desconhecida. Além disso, este estudo avaliou a contribuição do CGH‐array como uma ferramenta de diagnóstico de primeira linha em neonatos com anomalias congênitas avaliados por um programa de monitoramento de defeitos congênitos em uma maternidade pública no Sul do Brasil.
Até o momento, a maior triagem de recém‐nascidos (em 20.126 casos não selecionados) com análise de CGH‐array como teste de primeira linha revelou que 87/20.126 (0,43%) dos casos neonatais apresentaram desequilíbrios cromossômicos (53 casos de aneuploidias, 23 deleções e 11 duplicações).4
Reddy et al.14 relataram os resultados de um estudo de base populacional de 532 natimortos. Nessa amostra, a análise de CGH‐array gerou mais resultados que a análise do cariótipo (87,4% em comparação a 70,5%), proporcionou melhor detecção de anomalias genéticas (aneuploidia ou CNVs patogênicas, 8,3% em comparação a 5,8%) e também identificou mais desequilíbrios genômicos dentre 67 natimortos com anomalias congênitas (29,9% em comparação a 19,4%).
Na realização da análise seletiva, o uso de tecnologias com base em array demonstra a capacidade de detectar desequilíbrios patogênicos em aproximadamente 14‐18% dos casos pós‐natais com atrasos no desenvolvimento, deficiências intelectuais e ACMs encaminhados para análise.2,15–18 Nosso estudo verificou desequilíbrios genômicos em 4/35 (14,3%) dos casos que poderiam ser definidos como patogênicos e relacionados causalmente a fenótipos anormais. Embora nosso estudo tenha sido realizado em uma coorte relativamente pequena, a taxa de achados positivos detectados por meio do CGH‐array está dentro do intervalo relatado em diversas séries pós‐natais.
Embora exista uma clara associação entre CNVs em ambas as anomalias congênitas sindrômicas e não sindrômicas, apenas alguns grandes estudos de coorte realizaram especificamente uma análise de CGH‐array de todo o genoma em amostras de neonatos com defeitos congênitos. Lu et al.19 relataram a frequência de desequilíbrios genômicos identificados em 638 neonatos com vários defeitos congênitos encaminhados para análise cromossômica em microarranjos. Três diferentes plataformas de array com cobertura genômica cada vez mais ampla foram usadas, e os resultados obtidos foram comparados. Em geral, 17,1% dos pacientes foram identificados com anomalias clinicamente significativas, com taxas de detecção de 13,7%, 16,6% e 19,9%, dependendo da plataforma de array usada.
Neste estudo, uma análise de cariótipo anterior estava disponível em 32 casos e mostrou que a frequência de desequilíbrios cromossômicos detectada foi de 1/32 (3,1%). A taxa de detecção dos desequilíbrios genômicos não detectados anteriormente pela análise do cariótipo aumentou para 9/32 dos casos (28%) com o uso de CGH‐array, que estava de acordo com a expectativa de aumento da taxa de detecção. Em 4/35 (11,4%) dos casos, as CNVs poderiam ser definidas como patogênicas e relacionadas causalmente a fenótipos anormais. As diferenças de taxas entre diferentes estudos podem ocorrer devido ao tamanho da coorte, às diferenças na resolução da plataforma de array usada, aos critérios para a seleção de pacientes e à interpretação da relevância clínica das CNVs.
Dentre os 4/35 casos patogênicos, em dois deles (31 e 48), as anomalias identificadas encontradas não estavam previamente relacionadas a síndromes bem reconhecidas. Nos outros dois casos (1 e 14), alterações causais já tinham sido relacionadas a síndromes de microdeleção bem definidas (Síndrome de Wolf‐Hirschhorn (SWH) e Síndrome de Miller‐Dieker (SMD), respectivamente).20 Nesses dois casos com as CNVs relacionadas a disfunções genéticas bem definidas, os desequilíbrios cromossômicos poderiam ter sido previamente diagnosticados pela análise de cariótipo ou apenas por análise FISH (usando sondas específicas de um locus de uma região fundamental de um cromossomo) se os achados clínicos no momento do encaminhamento indicassem uma síndrome de microdeleção específica que pudesse informar exatamente qual(is) região(ões) e/ou cromossomo(s) investigar. Entretanto, ambas as amostras foram de indivíduos cujo cariótipo ou resultado da análise FISH não estavam disponíveis. Algumas disfunções genéticas, como a SWH e a SMD, são síndromes de microdeleção com CNV de tamanho variável famosas por serem causadas por genes sensíveis à dosagem, e as síndromes atípicas reconhecidas relacionadas a microdeleções não recorrentes podem não ser clinicamente diagnosticadas no nascimento. Adicionalmente, mesmo em uma síndrome bem definida, deleções cromossômicas não recorrentes podem ser de diferentes tamanhos, levando a um amplo espectro fenotípico.
Um dos dois casos com artrogripose múltipla congênita (caso 31) mostrou uma grande duplicação do braço longo do cromossomo 6 nas bandas q22.31‐q27 e uma deleção menor do braço longo do cromossomo 1 na banda q44. A recuperação dos registros laboratoriais mostrou que uma anomalia cromossômica de origem desconhecida envolvendo o braço longo do cromossomo 6 foi registrada anteriormente, porém não foi identificado qualquer desequilíbrio cromossômico envolvendo o cromossomo 1. Nesse momento, houve a expectativa de que os cariótipos dos pais seriam apresentados para melhor definir o tipo e origem (se de novo ou familiar) do material extra no cromossomo 6. A análise do CGH‐Array possibilitou a detecção de informações genômicas adicionais sobre a duplicação no cromossomo 6 identificada anteriormente e a detecção de um desequilíbrio genômico adicional (deleção no cromossomo 1) não relatado previamente. Frequentemente, mais de uma CNV é identificada em um indivíduo. Já é evidente, a partir da análise do cariótipo, que as duplicações cromossômicas devem envolver muitos genes e estar relacionadas causalmente às anomalias congênitas, conforme pressuposto no caso 31. Contudo, tem sido reconhecido que a presença de outra CNV poderia reduzir ou agravar o fenótipo clínico.21,22
Das duas amostras com fissura labial e/ou fissura palatina sindrômica (casos 14 e 34) e três amostras com fissura labial e palatina não sindrômica (casos 25, 30 e 37), um caso (14) apresentou uma deleção clinicamente significativa de 7,2 Mb no cromossomo na região 17p13.3‐p13.1, que coincide com a conhecida síndrome de microdeleção (SMD). Nos outros quatro casos, foram identificadas CNVs benignas (30 e 34) ou VOUS (casos 25 e 37). Aproximadamente 30% dos casos de fissura labiopalatina e 50% dos casos de fissura palatina foram reconhecidos como elementos de síndromes de ACM.23 Entretanto, fatores genéticos e ambientais são conhecidos por sua contribuição à ocorrência de fissuras labiopalatinas, complicando o esclarecimento dos mecanismos causais. Foram feitos esforços consideráveis na busca por gene(s) candidato(s) a fissuras não sindrômicas por meio do CGH‐array, mostrando ser um método eficaz para isolar os loci candidatos.24,25
A relevância clínica de 5/13 (36,7%) das CNVs entre os 12 casos com desequilíbrios genômicos continua incerta no momento, já que não há evidências suficientes para concluir se as CNVs eram patogênicas ou benignas. Quando são detectadas CNVs sem nenhum histórico ótimo de importância clínica, a interpretação da relação de causalidade entre as mesmas e defeitos congênitos pode ser desafiadora. Também devemos considerar que a CNV é possivelmente herdada de um pai saudável e, nesse caso, poderia ser uma variação patogênica com penetração incompleta ou uma alteração familiar benigna. A natureza altamente variável do genoma significa que é preciso tomar cuidado ao atribuir a patogenicidade às CNVs detectadas pelo CGH‐array. A partir das CNVs classificadas neste estudo como VOUS, podemos esperar que sejam realizados estudos dos pais para permitir uma melhor interpretação e fornecer informações valiosas ao aconselhamento genético antes de uma futura gravidez. De fato, é importante divulgar os dados referentes aos desequilíbrios cromossômicos com relevância clínica incerta, pois alguns dos dados podem representar CNVs recorrentes que poderiam estar relacionadas a novas síndromes. Relatos de pacientes com desequilíbrios genômicos e achados clínicos semelhantes podem levar à identificação de doenças genômicas recém‐reconhecidas ou genes candidatos relacionados a anomalias congênitas isoladas.
Em quatro casos (16, 30, 34 e 38), foram detectadas variantes normais classificadas como benignas. Sabe‐se que todos os seres humanos diferem em seus cromossomos no nível submicroscópico e que mesmo o genoma de indivíduos normais e saudáveis possui um elevado número de variações no número de cópias.26 Quando diversos indivíduos foram examinados para encontrar CNVs, foi identificado um total de 1.447 regiões com variações no número de cópias, abrangendo 360 Mb (12% do genoma).27 As CNVs são, com frequência, relativamente pequenas, podem ser herdadas de um genitor fenotipicamente normal, ocorrem em regiões cromossômicas com genes mais esparsos e contêm sequências de DNA mais repetitivas. A detecção de CNVs benignas foi relatada neste estudo mesmo em regiões genômicas que abrigam sistematicamente variantes benignas; isso pode reduzir a necessidade de realizar estudos dos pais de neonatos nos quais foram identificadas CNVs comprovadamente benignas.
Uma limitação a este estudo foi a incapacidade de diferenciar desequilíbrios genômicos de novo de desequilíbrios genômicos herdados devido à indisponibilidade de DNA dos pais. CNVs de novo em regiões do gene clinicamente significativas são mais propensas a serem causais. Entretanto, CNVs patogênicas herdadas não devem ser excluídas como causa de anomalias congênitas devido à sua expressividade variável e penetrância incompleta.28,29 CNVs patogênicas podem ser herdadas de um pai aparentemente normal e contribuir para o fenótipo anormal na criança. Esses tipos de CNVs são considerados loci de susceptibilidade, de forma que aumentam a chance de uma criança desenvolver anomalias congênitas, porém podem não ser suficientes para causar um fenótipo sozinhas. Estudos dos pais devem ser recomendados para indivíduos cujos achados clinicamente significativos foram relatados a fim de determinar se os achados de CNV representam eventos de novo ou familiares. Em casos de um desequilíbrio cromossômico de novo, também é recomendável o cariótipo dos pais para excluir uma translocação equilibrada em um dos pais. Embora diversas estratégias comuns tenham sido propostas para ajudar a interpretar os achados dos desequilíbrios genômicos,29,30 não existem critérios universais até agora. É essencial apresentar as informações mais precisas e atualizadas sobre a relevância clínica dos desequilíbrios genômicos detectados, bem como as CNVs em diferentes posições no genoma, mutações patogênicas ou polimorfismos em outros genes individuais ou causas não genéticas que possam ser necessárias para a expressão de uma anomalia congênita. É preciso ter cuidado na interpretação clínica dos resultados do CGH‐array. Poderão ser necessárias consultas adicionais em clínicas genéticas e uma análise estendida de familiares para prestar um aconselhamento preciso às famílias e calcular os riscos de recidiva.
Uma típica fragilidade dos estudos retrospectivos é a limitação das informações clínicas disponíveis. Obtivemos as informações clínicas disponíveis dos registros hospitalares durante o primeiro encaminhamento. A maioria dessas informações foi registrada no momento das exigências laboratoriais e eram, portanto, preliminares. Deve‐se salientar que 6/35 (17%) dos neonatos com anomalias congênitas morreram logo após o nascimento. Contudo, consideramos essa coorte representante dos neonatos em que a presença de desequilíbrios cromossômicos era suspeita.
Com este estudo, demonstramos a viabilidade e utilidade do CGH‐array na identificação de deleções e duplicações nas amostras de DNA armazenadas. Mostrou‐se que uma proporção de neonatos com anomalias congênitas de causa desconhecida apresentou desequilíbrios cromossômicos relacionados a seus fenótipos. Adicionalmente, este estudo demonstrou a detecção de anomalias cromossômicas compatíveis com síndromes genéticas em uma idade precoce, quando, normalmente, apenas alguns achados clínicos são claros.
Concluindo, o CGH‐array retrospectivo ou prospectivo como uma ferramenta de diagnóstico de primeira linha beneficiaria as famílias proporcionando um diagnóstico mais preciso e afetaria a gestão geral dos programas de monitoramento de defeitos congênitos em um número significativo de casos.
FinanciamentoCNPq/Brasil, bolsa 402012/2010‐0.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Os autores gostariam de agradecer o Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo apoio financeiro (bolsa 402012/2010‐0).
Como citar este artigo: Dorfman LE, Leite JC, Giugliani R, Riegel M. Microarray‐based comparative genomic hybridization analysis in neonates with congenital anomalies: detection of chromosomal imbalances. J Pediatr (Rio J). 2015;91:59–67.




