


To identify chromosomal imbalances by whole-genome microarray-based comparative genomic hybridization (array-CGH) in DNA samples of neonates with congenital anomalies of unknown cause from a birth defects monitoring program at a public maternity hospital.
MethodsA blind genomic analysis was performed retrospectively in 35 stored DNA samples of neonates born between July of 2011 and December of 2012. All potential DNA copy number variations detected (CNVs) were matched with those reported in public genomic databases, and their clinical significance was evaluated.
ResultsOut of a total of 35 samples tested, 13 genomic imbalances were detected in 12/35 cases (34.3%). In 4/35 cases (11.4%), chromosomal imbalances could be defined as pathogenic; in 5/35 (14.3%) cases, DNA CNVs of uncertain clinical significance were identified; and in 4/35 cases (11.4%), normal variants were detected. Among the four cases with results considered causally related to the clinical findings, two of the four (50%) showed causative alterations already associated with well-defined microdeletion syndromes. In two of the four samples (50%), the chromosomal imbalances found, although predicted as pathogenic, had not been previously associated with recognized clinical entities.
ConclusionsArray-CGH analysis allowed for a higher rate of detection of chromosomal anomalies, and this determination is especially valuable in neonates with congenital anomalies of unknown etiology, or in cases in which karyotype results cannot be obtained. Moreover, although the interpretation of the results must be refined, this method is a robust and precise tool that can be used in the first-line investigation of congenital anomalies, and should be considered for prospective/retrospective analyses of DNA samples by birth defect monitoring programs.
Identificar desequilíbrios cromossômicos por meio da hibridização genômica comparativa baseada em microarranjos (CGH-array) em amostras de DNA de neonatos com anomalias congênitas de causa desconhecida de um programa de monitoramento de defeitos congênitos em uma maternidade pública.
MétodosUma análise genômica cega foi realizada retrospectivamente em 35 amostras armazenadas de DNA de neonatos nascidos entre julho de 2011 e dezembro de 2012. Todas as possíveis variações no número de cópias (CNVs) de DNA foram comparadas com as relatadas em bases de dados genômicos públicas, e sua relevância clínica foi avaliada.
ResultadosDe um total de 35 amostras testadas, foram detectados 13 desequilíbrios genômicos em 12/35 casos (34,3%). Em 4/35 casos (11,4%), os desequilíbrios cromossômicos poderiam ser definidos como patogênicos; em 5/35 (14,3%) deles foram identificadas CNVs de DNA de relevância clínica incerta; e, em 4/35 (11,4%), foram detectadas variações normais. Dentre os quatro casos com resultados considerados relacionados causalmente aos achados clínicos, 2/4 (50%) apresentaram alterações causais já relacionadas a síndromes de microdeleção bem definidas. Em 2/4 amostras (50%), os desequilíbrios cromossômicos encontrados, embora preditivos como patogênicos, não estavam relacionados anteriormente a entidades clínicas reconhecidas.
ConclusõesA análise de CGH-array permitiu maior taxa de detecção de anomalias cromossômicas, e essa determinação é valiosa principalmente em neonatos com anomalias congênitas de etiologia desconhecida ou em casos em que os resultados do cariótipo não podem ser obtidos. Além disso, embora a interpretação dos resultados deva ser refinada, esse método é uma ferramenta robusta e precisa que pode ser usada na investigação de primeira linha de anomalias congênitas e deve ser considerada em análises futuras/retrospectivas de amostras de DNA por programas de monitoramento de defeitos congênitos.
Although Mendelian, chromosomal, and environmental causes have been established for many congenital anomalies and dysmorphic syndromes, the precise etiology of several such conditions has not yet been identified. Etiologic investigations of congenital anomalies suggest that 6% of birth defects are related to chromosomal abnormalities.1 However, the proportion of chromosomal anomalies in birth defects may be higher. Some individuals with congenital anomalies may have genomic imbalances below the resolution (> 5 Mb) of standard chromosome analysis. In the last decade, significant developments in the molecular detection of chromosomal imbalances have occurred, and their causal relationship to congenital anomalies and mental disabilities has increased. The considerable gap between the resolution for detection of chromosome abnormalities with light microscopy and molecular gene analysis was bridged with the introduction of molecular approaches, such as microarray-based comparative genomic hybridization (array-CGH). Array-CGH is currently a powerful method for the simultaneous detection of chromosomal imbalances and the most prevalent chromosome abnormalities. It allows for the detection of trisomies and large chromosomal anomalies (already recognized by standard karyotype analysis) as well as smaller submicroscopic chromosomal imbalances (deletions, duplications, or triplications of any chromosomal region, few of which are recognized by fluorescence in situ hybridization [FISH]) that result in copy-number variations (CNVs). Several studies have shown that the use of array-based technologies increases the detection rate of chromosomal abnormalities to approximately 14% to 18%, compared with a rate of approximately 3% (excluding trisomy 21) using standard cytogenetic approaches in individuals with developmental delays, intellectual disabilities, learning difficulties, multiple congenital abnormalities (MCAs), autistic spectrum disorders, schizophrenia, and other neuropsychiatric disorders.2 The overall frequency of unbalanced chromosome abnormalities was reported in neonates as 0.43%, according to recent reports.3,4 Therefore, the introduction of genome-wide array-CGH analysis at the neonatal period, when few clinical findings related to recognized causes may be present, potentially increases the possibility of early detection of chromosomal abnormalities consistent with a genetic/genomic disorder.
Therefore, the aim of this study was to identify chromosomal imbalances using a retrospective whole-genome array-CGH analysis in stored DNA samples of neonates with congenital anomalies of unknown cause. In addition, this study evaluated the contribution of array-CGH as a first-line diagnostic tool in neonates with congenital anomalies evaluated by a birth defects monitoring program at a public maternity hospital in Southern Brazil.
MethodsSample selectionThis retrospective study was performed using de-identified DNA samples extracted from the blood of neonates, which were obtained from the biorepository of the Programa de Monitoramento de Defeitos Congênitos (PMDC) of Hospital de Clínicas de Porto Alegre (HCPA), Brazil. Subjects were less than 30 days of age, presenting a wide range of congenital anomalies of unknown cause and in whom a chromosomal abnormality was suspected. The clinical indications for cytogenetic analysis at the time of referral were taken from the clinical and laboratory data collected at birth and available in hospital records, and did not include follow-up investigations and information about disease outcomes. Cases without enough clinical data were excluded, as were cases where mothers had clinical or laboratory suspicion of infectious/parasitic diseases or a history of use/abuse of illicit drugs/alcohol during pregnancy. According to these criteria, a total of 45 samples were selected, but ten were excluded because they did not achieve the optimal DNA quality needed for the array-CGH analysis, and thus, the study was conducted with 35 samples. The results of previous chromosome analyses were obtained in 32 cases. Conventional cytogenetic testing at the 500-550 band level resolution was initially normal for all cases, but in one case a report of an abnormal karyotype was provided later. This study was approved by the Institutional Review Board of the HCPA and was conducted in accordance with current institutional ethics rules regarding the use of biological materials from biorepositories.5
Whole-genome Array-CGHOligonucleotide array-based CGH was performed using an 8×60k whole-genome platform (design 021924, Agilent Technologies, Santa Clara, USA), which has an average spacing of 40kb between probes. Genomic DNA was isolated from the peripheral blood (provided by the PMDC-HCPA) of 35 neonatal individuals and subsequently analyzed. For each experiment, a gender-mismatched normal reference (Promega Corp. Madison, WI, USA) was used. The experiments were performed according to the manufacturer's protocol. Images of the arrays were taken using a microarray scanner (design G2600D, Agilent, California, USA) and processed using Feature Extraction software (design v9.5.1, Agilent, California, USA), both from Agilent. The raw data were analyzed by Agilent Cytogenomics (design v2.7.8.0, Agilent, California, USA) software with the statistical algorithm ADM-2, using a threshold of 6.0 and a four-probe minimum aberration call. Subsequent software normalization of the data was performed for the verification of DNA copy number changes. The p-values for each probe were calculated, providing additional objective statistical criteria to determine whether the deviation of each probe from zero was statistically significant.6 All experiments included two array hybridizations per sample, and the results were recorded and compared. Only genomic imbalances detected in both dye-swap experiments were reported.
Data AnalysisWhole-genome array-CGH data analyses were performed in a blinded fashion; samples were received, de-identified, and investigators who performed the array-CGH analyses were not aware of the previous clinical and laboratory information related to each sample. The DNA copy number variations (CNVs) detected were compared with CNVs reported in publicly available online resources and databases of chromosomal abnormalities and variants.7–13 The CNVs (gains/duplications or losses/deletions) were classified into different categories: benign CNV (normal genomic variant); CNV of uncertain clinical relevance (variant of uncertain significance [VOUS]); and CNV of possible clinical relevance (pathogenic variant). In this study, the pathogenic abnormalities included the detection of CNVs in known pathogenic regions, deletion/duplication > 3 Mb in size, or visible by G-banded karyotype that have not been reported in the normal population, and deletions or duplications < 3 Mb previously reported as pathogenic. Benign deletions or duplications included variants well documented in the normal population or previously reported as benign. Deletions or duplications were classified as being VOUS when insufficient evidence was available to conclude if the CNV was either pathogenic or benign.
ResultsThe data of the 35 neonates with congenital anomalies of unknown cause, born between July of 2011 and December of 2012, whose DNA samples were analyzed by whole-genome array-CGH, are presented in Table 1. The maternal age ranged from 16 to 41 years of age. This study identified 12 (34.3%) cases with DNA copy number variations (CNVs). From those cases, 7/12 (58.3%) were male and 5/12 (41.7%) were female. The details of the array-CGH results from the cases with genomic imbalances are summarized in Table 2. Thirteen CNVs were identified in 12 individuals. Overall, duplications were verified in 6/35 (17%) and deletions were verified in 7/35 (20%) of the cases. In 6/35 (17%) of the cases, only a deletion was identified; 5/35 cases (14.3%) only had a duplication, and 1/35 (2.8%) had a deletion and a duplication. Additionally, a FISH test confirmed the array-CGH results in one deletion case (case 14) from which stored cells were available (data not shown).
Summary of the clinical indications from the 35 samples at the time of referral for chromosomal analysis.
| Case | Main clinical associated features |
|---|---|
| 1a | Female, CDH, microtia, hypertelorism, CHD (dextroposition of the heart), anal atresia |
| 2 | Male, CHD (tetralogy of Fallot) |
| 4 | Male, omphalocele, microcephaly |
| 5 | Male, omphalocele, limb agenesis, ambiguous genitalia, anal atresia, bladder dysfunction |
| 10 | Female, gastroschisis |
| 12a | Male, CDH |
| 13 | Male, unilateral phocomelia, hip dysplasia |
| 14 | Male, cranial asymmetry, cleft palate (soft), ocular hypertelorism, esophageal atresia type IIIB, camptodactyly of the 3rd, 4th, and 5th fingers, clinodactyly of the 5th finger, clubfeet |
| 15 | Male, anal atresia, club feet |
| 16 | Female, anal atresia, hypoplastic genitalia, CHD |
| 17 | Female, arthrogryposis multiplex congenital (amyoplasia) |
| 19 | Female, non-syndromic unilateral cleft lip (left) and cleft palate |
| 22a | Female, microcephaly, cerebellar hypoplasia, olygohydramnios, pulmonary hypoplasia, renal dysplasia, genital hypoplasia, frontal microgyria, occipital encephalocoele, cerebellar hypoplasia |
| 23 | Female, omphalocele, microcephaly |
| 24 | Male, micrognathia, single upper median incisor |
| 25 | Female, non-syndromic cleft lip and palate (bilateral) |
| 29 | Male, CHD (tetralogy of Fallot) |
| 30 | Male, non-syndromic cleft lip and palate (bilateral) |
| 31a | Male, arthrogryposis multiplex congenita (amyoplasia), CHD (tetralogy of Fallot), hip dysplasia |
| 32a | Female, bilateral multicystic dysplastic kidney, oligohydramnios |
| 33 | Male, cleft palate, clubfeet |
| 34 | Male, cleft lip (left) and cleft palate, widow's peak, widely spaced nipples, genital hypoplasia, hypospadias |
| 35 | Female, HPE, oligohydramnios, microcephaly, unilateral choanal atresia |
| 37 | Male, non-syndromic cleft lip and palate (bilateral) |
| 38 | Male, esophageal atresia type IIIB |
| 40 | Male, intrauterine growth retardation |
| 41 | Female, gastrosquisis |
| 42 | Female, CHD (tetralogy of Fallot) |
| 43 | Male, non-syndromic cleft palate |
| 44 | Male, microgyria, incomplete lissencephaly, micrognathia |
| 46 | Male, meningocele, club foot (left) |
| 47 | Female, gastroschisis |
| 48a | Male, bilateral multicystic dysplastic kidney, oligohydramnios, bilateral pulmonary hypoplasia |
| 49 | Female, gastroschisis |
| 50 | Male, arthrogryposis multiplex congenita, micrognathia |
CDH, congenital diaphragmatic hernia; CHD, congenital heart defect; HPE, holoprosencephaly.
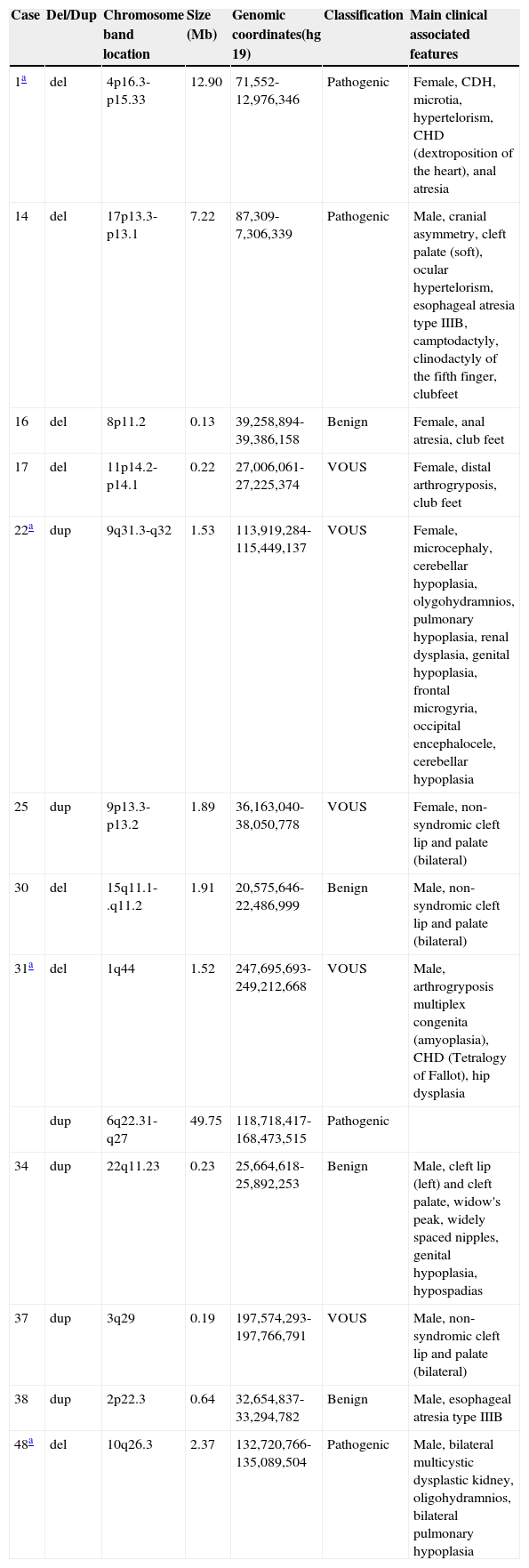
Details of the array-CGH from 12 samples with chromosomal imbalances.
| Case | Del/Dup | Chromosome band location | Size (Mb) | Genomic coordinates(hg 19) | Classification | Main clinical associated features |
|---|---|---|---|---|---|---|
| 1a | del | 4p16.3-p15.33 | 12.90 | 71,552-12,976,346 | Pathogenic | Female, CDH, microtia, hypertelorism, CHD (dextroposition of the heart), anal atresia |
| 14 | del | 17p13.3-p13.1 | 7.22 | 87,309-7,306,339 | Pathogenic | Male, cranial asymmetry, cleft palate (soft), ocular hypertelorism, esophageal atresia type IIIB, camptodactyly, clinodactyly of the fifth finger, clubfeet |
| 16 | del | 8p11.2 | 0.13 | 39,258,894-39,386,158 | Benign | Female, anal atresia, club feet |
| 17 | del | 11p14.2-p14.1 | 0.22 | 27,006,061-27,225,374 | VOUS | Female, distal arthrogryposis, club feet |
| 22a | dup | 9q31.3-q32 | 1.53 | 113,919,284-115,449,137 | VOUS | Female, microcephaly, cerebellar hypoplasia, olygohydramnios, pulmonary hypoplasia, renal dysplasia, genital hypoplasia, frontal microgyria, occipital encephalocele, cerebellar hypoplasia |
| 25 | dup | 9p13.3-p13.2 | 1.89 | 36,163,040-38,050,778 | VOUS | Female, non-syndromic cleft lip and palate (bilateral) |
| 30 | del | 15q11.1-.q11.2 | 1.91 | 20,575,646-22,486,999 | Benign | Male, non-syndromic cleft lip and palate (bilateral) |
| 31a | del | 1q44 | 1.52 | 247,695,693-249,212,668 | VOUS | Male, arthrogryposis multiplex congenita (amyoplasia), CHD (Tetralogy of Fallot), hip dysplasia |
| dup | 6q22.31-q27 | 49.75 | 118,718,417-168,473,515 | Pathogenic | ||
| 34 | dup | 22q11.23 | 0.23 | 25,664,618-25,892,253 | Benign | Male, cleft lip (left) and cleft palate, widow's peak, widely spaced nipples, genital hypoplasia, hypospadias |
| 37 | dup | 3q29 | 0.19 | 197,574,293-197,766,791 | VOUS | Male, non-syndromic cleft lip and palate (bilateral) |
| 38 | dup | 2p22.3 | 0.64 | 32,654,837-33,294,782 | Benign | Male, esophageal atresia type IIIB |
| 48a | del | 10q26.3 | 2.37 | 132,720,766-135,089,504 | Pathogenic | Male, bilateral multicystic dysplastic kidney, oligohydramnios, bilateral pulmonary hypoplasia |
CDH, congenital diaphragmatic hernia; CHD, congenital heart defect; VOUS, variant of uncertain significance; array-CGH, microarray-based comparative genomic hybridization; del, deletion; dup, duplication.
Among the five individuals with syndromic or non-syndromic orofacial clefts (cases 14, 25, 30, 34, and 37) in whom genomic imbalances were detected, one case exhibited a clinically significant 7.2 Mb deletion at chromosome 17p13.3-p13.1 (case 14) that coincides with the known Miller-Dieker syndrome (MDS) region. FISH analysis confirmed the deletion of the chromosome 17p13.3 region (data not shown). The other four cases with oral facial clefts showed CNVs that were classified as benign or as VOUS. Of the two cases with arthrogryposis multiplex congenital (17 and 31), in one individual an interstitial duplication of the long arm of chromosome 6 at band q22.31-q27 was found, as well as a terminal deletion of the long arm of chromosome 1 at band q44. The previous karyotype analysis showed the identification of a chromosomal abnormality of unknown origin involving the long arm of chromosome 6, but not the chromosomal imbalance that involved chromosome 1. This infant died at the age of 35 days. Of five additional cases with MCAs (1, 16, 22, 38, and 48), this study identified clinically significant chromosomal imbalances or potential pathogenic CNVs in three cases (1, 31, and 48). The subjects died at the age of 2 days, 5hours, and 3 days after birth, respectively. Overall, the deletions were classified as pathogenic in three cases (1, 14, and 48), as benign in two cases (16 and 30), and as VOUS in two cases (17 and 31). The duplications were classified as pathogenic in one case (31), as benign in two cases (34 and 38), and as VOUS in three cases (cases 22, 25, and 37). Examples of array-CGH graphical overviews are shown in Fig. 1.
 and DNA from normal subjects as reference (in blue). The test/reference ratio data for each chromosome are shown. Each dot represents a single probe (oligo) spotted on the array. The log ratio of the chromosome probes is plotted as a function of chromosomal position. Copy number loss shifts the ratio to the left (value of approximately -1x). Copy number gain shifts the ratio to the right (value of approximately +1x). The ideogram of each chromosome (left margin) shows the location of each probe. The probe log2 ratios were plotted according to genomic coordinates (based on the UCSC Genome Browser, February 2009, NCBI Build 37 reference sequence). A: A ∼1.5 Mb terminal deletion at chromosome 1q44 (blue line) in case 31. B: A ∼12.9 Mb terminal deletion at chromosome 4p16.3-p15.33 (blue box) in case 1. C: A ∼ 49.7 Mb interstitial duplication at chromosome 6q22.31-q37 (blue box) in case 31. D: A ∼ 2.37 Mb terminal deletion at chromosome 10q26.3 (blue box) in case 48. E: A ∼ 7.2 Mb terminal deletion at chromosome 17p13.3-p13.1 (blue box) in case 14.")
Array-CGH ratio profiles of the chromosomes in four neonates with pathogenic chromosomal imbalances using genomic DNA from the neonates as test (in red) and DNA from normal subjects as reference (in blue). The test/reference ratio data for each chromosome are shown. Each dot represents a single probe (oligo) spotted on the array. The log ratio of the chromosome probes is plotted as a function of chromosomal position. Copy number loss shifts the ratio to the left (value of approximately -1x). Copy number gain shifts the ratio to the right (value of approximately +1x). The ideogram of each chromosome (left margin) shows the location of each probe. The probe log2 ratios were plotted according to genomic coordinates (based on the UCSC Genome Browser, February 2009, NCBI Build 37 reference sequence). A: A ∼1.5 Mb terminal deletion at chromosome 1q44 (blue line) in case 31. B: A ∼12.9 Mb terminal deletion at chromosome 4p16.3-p15.33 (blue box) in case 1. C: A ∼ 49.7 Mb interstitial duplication at chromosome 6q22.31-q37 (blue box) in case 31. D: A ∼ 2.37 Mb terminal deletion at chromosome 10q26.3 (blue box) in case 48. E: A ∼ 7.2 Mb terminal deletion at chromosome 17p13.3-p13.1 (blue box) in case 14.
The aim of this study was to retrospectively identify genomic imbalances using whole-genome array-CGH in samples available from neonates with congenital anomalies of unknown etiology. In addition, this study evaluated the contribution of array-CGH as a first-line diagnostic tool in neonates with congenital anomalies in a birth defects monitoring program at a public maternity hospital in Southern Brazil.
To date, the largest newborn screening (in 20,126 unselected cases) using array-CGH analysis as a first-line test revealed that 87/20,126 (0.43%) of the neonatal cases had chromosomal imbalances (53 cases of aneuploidies, 23 deletions, and 11 duplications).4
Reddy et al.14 reported the results of a population-based study of 532 stillbirths. In this sample, array-CGH analysis yielded more results than did karyotype analysis (87.4% vs. 70.5%), provided better detection of genetic abnormalities (aneuploidy or pathogenic CNVs, 8.3% vs. 5.8%), and also identified more genomic imbalances among 67 stillbirths with congenital anomalies (29.9% vs. 19.4%).
When selective screening is performed, the use of array-based technologies demonstrates the ability to detect pathogenic imbalances in approximately 14%-18% of postnatal cases with developmental delays, intellectual disabilities, and MCAs referred for analysis.2,15–18 The present study verified genomic imbalances in 4/35 (14.3%) of the cases that could be defined as pathogenic and causally related to the abnormal phenotypes. Although this study was performed in a relatively small cohort, the rate of positive findings detected through array-CGH is in the range reported in several postnatal series.
Although a clear association exists between CNVs in both syndromic and non-syndromic congenital anomalies, only few large cohort studies have specifically performed whole-genome array-CGH analysis in samples of neonates with birth defects. Lu et al.19 reported the frequency of genomic imbalances identified in 638 neonates with various birth defects referred for chromosomal microarray analysis. They used three different array platforms with increasingly extensive genomic coverage and compared the results obtained. Overall, 17.1% of patients were identified with clinically significant abnormalities, with detection rates of 13.7%, 16.6%, and 19.9%, depending on the array platform used.
In the present study, a previous karyotype analysis was available in 32 cases and showed that the frequency of chromosomal imbalances detected was 1/32 (3.1%). The detection yield of genomic imbalances not previously detected by karyotype analysis increased to 9/32 cases (28%) with the use of array-CGH, which was in agreement with the expected increased detection yield. In 4/35 cases (11.4%), CNVs could be defined as pathogenic and causally related to the abnormal phenotypes. Rate differences between different studies may be due to the cohort size, differences in the resolution of the array platform used, the criteria for patient selection, and the interpretation of the clinical relevance of the CNVs.
Among the 4/35 pathogenic cases, in two cases (31 and 48), the identified abnormalities found had not been previously associated with well-recognized syndromes. In the two other cases (1 and 14), causative alterations had already been associated with well-defined microdeletion syndromes20 (Wolf-Hirschhorn Syndrome [WHS] and MDS, respectively). In these two cases with CNVs associated with well-defined genetic disorders, the chromosomal imbalances could have been previously diagnosed by karyotype analysis or by FISH analysis alone (using locus-specific probes for the critical chromosome region) if the clinical findings at the time of referral were indicative of a particular microdeletion syndrome that could inform exactly which region(s) and/or chromosome(s) to investigate. However, both samples were from subjects in whom neither karyotype nor FISH analysis results were available. Certain genetic disorders, such as WHS and MDS, are microdeletion syndromes with CNV of variable size known to be caused by dosage-sensitive genes, and atypical recognized syndromes associated with non-recurrent microdeletions might be clinically missed at birth. Furthermore, even in a well-defined syndrome, non-recurrent chromosome deletions can be of different sizes, leading to a broad phenotypic spectrum.
One of the two cases with arthrogryposis multiplex congenita (case 31) showed a large duplication of the long arm of chromosome 6 at bands q22.31-q27 and a smaller deletion of the long arm of chromosome 1 at band q44. The retrieval of laboratory records showed that a chromosomal abnormality of unknown origin involving the long arm of chromosome 6 was previously recorded, but no chromosomal imbalance involving chromosome 1 was identified. At that time, there was an expectation that parental karyotypes would be performed to better define the type and origin (de novo or familial) of the extra material on chromosome 6. Array-CGH analysis allowed for additional genomic information regarding the previously identified duplication at chromosome 6 and the detection of an additional genomic imbalance (deletion at chromosome 1) that was not previously reported. Frequently, more than one CNV is identified in an individual. It is evident already from the karyotype analysis that the chromosome duplications must involve many genes and be causally related to the congenital anomalies, as assumed in case 31. However, it has been recognized that the presence of another CNV could reduce or aggravate the clinical phenotype.21,22
From the two samples with syndromic cleft lip and/or cleft palate (cases 14 and 34) and the three with non-syndromic cleft lip and cleft palate (cases 25, 30, and 37), one case (14) had a clinically significant 7.2 Mb deletion at chromosome 17p13.3-p13.1 that coincides with the known MDS microdeletion syndrome. In the other four cases, benign CNVs (30 and 34) or VOUS were identified (cases 25 and 37). Approximately 30% of cleft lip and palate cases and 50% of cleft palate cases are recognized as components of MCA syndromes.23 However, both genetic and environmental factors are known to contribute to the occurrence of cleft lip and palate, complicating the elucidation of the causative mechanisms. Considerable efforts have been made in seeking candidate gene(s) for non-syndromic clefts through array-CGH, showing that it is an effective method for isolating candidate loci.24,25
The clinical relevance of 5/13 (36.7%) CNVs among the 12 cases with genomic imbalances remains uncertain at present, as there is insufficient evidence to conclude whether the CNVs were either pathogenic or benign. When CNVs are detected that have no strong track record for clinical importance, the interpretation of whether they are causal for the birth defect can be challenging. It should also be considered that the CNV is potentially inherited from a healthy parent and, in this case, could be a pathogenic variant with incomplete penetrance or a benign familiar variation. The highly variable nature of the genome means that care must be taken in assigning pathogenicity to CNVs detected by array-CGH. From the CNVs classified as VOUS in this study, it might be expected that parental studies would be performed to allow a better interpretation and to provide valuable information for genetic counseling prior to a future pregnancy. Indeed, it is important to report data on chromosome imbalances with unclear clinical significance, because some of the data may represent recurrent CNVs that could be associated with novel syndromes. Reports of patients with similar genomic imbalances and clinical findings can lead to the identification of newly recognized genomic disorders or candidate genes associated with isolated congenital anomalies.
In four cases (16, 30, 34, and 38), normal variants classified as benign were detected. It is recognized that all humans differ in their chromosomes at the submicroscopic level and that even the genomes of normal, healthy individuals have a high number of copy number changes.26 When several individuals were screened for CNVs, a total of 1,447 copy number variable regions covering 360 Mb (12% of genome) were identified.27 CNVs are often relatively small, can be inherited from a phenotypically normal parent, occur in more gene-sparse chromosomal regions, and contain more repetitive DNA sequences. The detection of benign CNVs was reported in this study from genomic regions that consistently harbor benign variants; this might reduce the need to perform parental studies in neonates in whom proven benign CNVs were identified.
A limitation of this study was the inability to distinguish de novo from inherited genomic imbalances due to the unavailability of parental DNA. De novo CNVs in clinically significant gene regions are more likely to be causative. However, inherited pathogenic CNVs should not be excluded as a cause of congenital anomalies because of their variable expressivity and incomplete penetrance.28,29 Pathogenic CNVs may be inherited from an apparently normal parent and contribute to the abnormal phenotype in the child. These types of CNVs are thought of as susceptibility loci, in that they increase the chance of a child developing congenital anomalies but may not be sufficient to cause a phenotype by themselves. Parental studies should be recommended for individuals for whom clinically significant findings were reported, to determine whether the CNV findings represent de novo or familiar events. In cases of a de novo chromosome imbalance, it is also recommended to obtain the parental karyotype in order to exclude a balanced translocation in one of the parents. Although several common strategies have been proposed to help interpret the findings of genomic imbalances,29,30 there are no universal criteria thus far. It is essential to have the most accurate and up-to-date information on the clinical significance of detected genomic imbalances, as well as CNVs at different positions in the genome, pathogenic mutations or polymorphisms in other individual genes, or nongenetic causes that might be required for a congenital anomaly to be expressed. Caution must be taken in the clinical interpretation of the array-CGH results. Further consultations at genetics clinics and extended analysis in family members may be necessary to provide accurate counseling to the families and to calculate the recurrence risks.
A typical weakness of retrospective studies is the limited clinical information available. The present study retrieved the clinical information available from the hospital records during the first referral. Most of these were recorded at the time of the first laboratory requirements and were therefore preliminary. Of note, 6/35 (17%) neonates with congenital anomalies died soon after birth. Nevertheless, the authors consider this cohort representative of the neonates in whom the presence of chromosomal imbalances was suspected.
This study demonstrated the feasibility and usefulness of array-CGH to identify deletions and duplications in stored DNA samples. It was shown that a proportion of neonates with congenital anomalies of unknown cause had chromosomal imbalances associated with their phenotypes. Furthermore, this study demonstrated the detection of chromosomal abnormalities consistent with genetic syndromes at an early age, when often, only a few clinical findings are clear.
In conclusion, retrospective or prospective array-CGH as a first-line diagnostic tool would benefit families by providing a more accurate diagnosis and impact the overall management in a significant number of cases from birth defects monitoring programs.
FundingCNPq/Brazil, grant 402012/2010-0.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support (grant 402012/2010-0).
Please cite this article as: Dorfman LE, Leite JC, Giugliani R, Riegel M. Microarray-based comparative genomic hybridization analysis in neonates with congenital anomalies: detection of chromosomal imbalances. J Pediatr (Rio J). 2015;91:59–67.




