

To provide an overview of drug treatment, transplantation, and gene therapy for patients with primary immunodeficiencies.
Source of dataNon-systematic review of the literature in the English language carried out at PubMed.
Synthesis of dataThe treatment of patients with primary immunodeficiencies aims to control their disease, especially the treatment and prevention of infections through antibiotic prophylaxis and/or immunoglobulin replacement therapy. In several diseases, it is possible to use specific medications for the affected pathway with control of the condition, especially in autoimmune or autoinflammatory processes associated with inborn immunity errors. In some diseases, treatment can be curative through hematopoietic stem cell transplantation (HSCT); more recently, gene therapy has opened new horizons through new technologies.
ConclusionsImmunoglobulin replacement therapy remains the main therapeutic tool. Precision medicine with specific drugs for altered immune pathways is already a reality for several immune defects. Advances in the management of HSCT and gene therapy have expanded the capacity for curative treatments in patients with primary immunodeficiencies.
Primary immunodeficiencies (PIDs), recently called inborn errors of immunity (IEI), are a growing group of more than 400 diseases, mostly of monogenic origin, associated with pathogenic variations of more than 430previously described genes. Patients with IEI have a wide spectrum of clinical manifestations, from mildly symptomatic patients with a late diagnosis to those with severe symptoms and risk of death, which is significant from the first months of life onwards.1
Most IEI lead to changes in essential immune pathways resulting in increased susceptibility to common and opportunistic pathogens; however, in other cases these changes in the immune system lead to greater susceptibility to a single microorganism or a restricted group of pathogens.2 In addition to infections, primary immunodeficiencies can occur together with changes in the immune system regulation, predisposing the patient to autoimmune and autoinflammatory diseases or severe allergies, which can develop as complications throughout life or even, in an IEI group, constitute the main clinical manifestations.3,4
The care and treatment of patients with IEI differ according to the result of the defect, the affected immune pathway, and the severity of each case. Therefore, an accurate diagnosis is crucial for adequate treatment, including general care, broad-spectrum or specific pharmacotherapies, such as the use of biological agents, and the use of curative therapies, such as bone marrow transplantation and gene therapy.5 The objective of this review is to provide an overview of the management of patients with IEI.
Antibiotic therapyInfections are the most common forms of presentation in patients with IEI and depend on the type of immune defect present. The treatment of infections in patients with IEI is complex, requiring long-term use of medication and often those with a broad spectrum. Due to the greater susceptibility to unusual agents, a greater effort must be made for the exact identification of pathogens, including the culture of affected tissues and molecular techniques to identify the pathogen.6
The use of prophylactic antibiotic therapy is quite widespread in the management of patients with primary immunodeficiencies, aiming to reduce the frequency and severity of infections, especially sinopulmonary infections caused by common bacteria; in some PIDs with more specific susceptibilities, prophylactic antiviral and/or antifungal therapy may be necessary.7 Most physicians treating patients with IEI reported the use of prophylactic antimicrobials in at least some of their patients.8 However, there is scarce scientifically-based evidence for the use of antibiotic prophylaxis in PIDs, except for chronic granulomatous disease (CGD) and severe combined immunodeficiency (SCID).9–11 Recently, the prophylactic use of azithromycin in patients with antibody defects (common variable immunodeficiency [CVID] and agammaglobulinemia) undergoing immunoglobulin replacement therapy showed a reduction in the number of annual exacerbations, use of antibiotics for treatment, and risk of hospitalization.12
In addition to the above mentioned PID pictures, isolated antibiotic prophylaxis is often offered to patients with mild hypogammaglobulinemia, selective immunoglobulin A (IgA) deficiency or deficiency of IgG subclasses, who are not receiving immunoglobulin, despite the lack of evidence to support the use of antibiotics in this population. In these cases, the medications are used at certain times of the year (especially in winter) or continuously, depending on the individual analysis of each case. A careful monitoring of these patients' adverse effects and infection rates is important.13
Table 1 presents several antibiotic prophylaxis regimens currently used in the treatment of patients with IEI, grouped in a very convenient way in a non-systematic review, and then translated and adapted to the Brazilian context. As there is no standardization or formal consensus, the agents and doses shown in the Table are those most commonly used; however, other regimens are used in different centers and may also be adequate.14
Examples of antibiotic prophylaxis regimens used in patients with immunodeficiency.
| Prevention intention | Preferential regimen | Alternative regimen |
|---|---|---|
| Pneumocystis jirovecii | Sulfametoxazol-trimetoprima (SXT-TMP):. Infants> 4 weeks of age and children:5mg/kg/day divided into two doses 3x/week(Based on TMP, maximum 160mg per day). Adults and adolescents with normal kidney function: based on TMP 80mg/day or 160mg daily or 160mg 3x/week | Dapsone:. Infants and children: 2mg/kg/day daily 1x/day (maximum: 100mg/day). Adults: 100mg 1x/day or 50mg 2x/dayPentamidine:. Children <5 years: 9mg/kg (maximum:300mg/dose) nebulized inhalation every 4weeks. Children> 5 years, adolescents and adults:300mg nebulized inhalation every 4 weeks |
| Staphylococcus spp, Gram-negative spp | SXT-TMP. Infants> 4 weeks of age and children:5mg/kg/day divided into 2 daily doses(Based on TMP, maximum 160mg per day)Adults and adolescents: based on TMP 160mg daily | Amoxicillin:a. Children: 10–20mg/kg per day, single dose or divided into 2x (maximum: 875mg/day). Adolescents and adults: 875mgCiprofloxacin:a,b#. Children: 10mg/kg/dose 2x/day (maximum: 500mg). Adults: 500mgAmoxicillin and clavulanate:a. Children: 20mg/kg per day single dose or divided into 2x (maximum: 875mg/day based on amoxicillin). Adolescents and adults: 875mg (based on amoxicillin) |
| Mycoplasma spp, Streptococcus spp | Azithromycin. Children: 5–10mg/kg/oral dose 3x/week(maximum: 250mg). Adolescents and adults: 250mg oral dose 3x/week | |
| Atypical mycobacteriosis | Azithromycin. Children: 20mg/kg/oral dose 1x/week(maximum dose of 1200mg/week; can be given up to 600mg 2x/without causing nausea at high doses). Adolescents and adults: 1200mg 1x/week (or 600 2x/week in case of nausea) | |
| Aspergillus spp | Itraconazole:. Children: 5mg/kg/day orally (maximum: 200mg). Adolescents and adults: 200mg oral daily | Voriconazolec:.≤50kg: 8mg/kg/oral dose 2x/day(maximum per dose: 350mg)> 50 kg: 4mg/kg/oral dose 2x/day(maximum per dose: 200mg) |
| Candida spp | Fluconazole:. Children: 6mg/kg orally daily (maximum: 400mg). Adolescents and adults: 400mg orally daily | |
| HSV/VZV | Acyclovir:. Children <40 kg: 600mg oral dose 4x/day. Children> 40 kg: 800mg oral dose 4 x/day. Adults: 800mg oral dose 2x/day | |
| CMV | Valganciclovir:. Children 1 month to 16 years: oral dose (mg)=7 × body surface area×creatinine clearance. Adolescents ≥17 years and adults with normal renal function: 900mg oral dose 1x/day |
spp, species; HSV, herpes simplex virus; VZV, varicella zoster virus; CMV, cytomegalovirus.
Adapted from Bundy et al.14
Around 50%–75% of IEI patients require immunoglobulin replacement, because antibody production is absent or inadequate. In turn, with the advances in the treatment of lymphomas, leukemias, and other types of cancer, the number of cases of secondary immunodeficiency that affect antibody production is increasing and must be remembered. In Brazil, we currently have medications for intravenous or subcutaneous administration. We will address the main indications and the comparison between intravenous and subcutaneous administration.
Regarding the therapeutic benefit, immunoglobulin replacement therapy indications can be classified as:
• Proven benefit
Immune system defects that affect B-cells
Hypogammaglobulinemia and inefficient antibody production
• Likely benefit
Immunoglobulins with apparently normal levels, but with a qualitative defect in the specific antibody production
• No benefit/Contraindicated
Selective IgA deficiency
IgG4 deficiency
From a practical standpoint, the recommendation of the European Immunodeficiency Society (ESID) can be followed for immunoglobulin replacement therapy indication:15
1) IgG < 200mg/dL: all patients
2) IgG 200−500mg/dL: associated with repeated infections
3) IgG > 500mg/dL: specific antibody deficiency associated with severe or repeated infections
The main immunodeficiencies that require immunoglobulin replacement therapy are:
• Agammaglobulinemia:
It refers to a defect in the ontogeny of B lymphocytes, which become absent and, therefore, there is no antibody production. This group of diseases can be detected in neonatal screening by the KRECs test and later confirmed by complete lymphocyte immunophenotyping.
• Hypogammaglobulinemia
In this case, there is a reduction in the production of antibodies and a decrease in the serum levels of immunoglobulins.16 The classic example of this group of diseases is CVID, which can be the result of several genetic alterations.
• Hyper-IgM syndrome
These diseases are characterized by reduced levels of IgA and IgG and normal or elevated levels of IgM. The number of B-lymphocytes is usually normal, but patients have a clinical picture of repeated infections similar to cases of agammaglobulinemia or combined immunodeficiency.17
• Antibody deficiency with normal levels of Immunoglobulins
Deficient response to polysaccharide antigens associated with severe infections and risk of sequelae.2,18
Patients with hyper-IgE syndrome usually have normal levels of immunoglobulins, but some have antibody production deficiency after immunization.19 In Wiskott-Aldrich syndrome, there is also impaired antibody production to protein and polysaccharide antigens, and immunoglobulin replacement helps in the reduction of infectious conditions until the transplantation is performed.20,21 In cases of ataxia-telangiectasia, a significant number of patients have repeated infections, as well as cell and humoral immunity alterations.22
There is no indication for immunoglobulin replacement in patients with selective IgA deficiency, unless there is an association with deficiency of IgG subclasses or a qualitative defect in antibody production.23 Immunoglobulin replacement should also be considered in cases of cancer, lymphoma, leukemias, and for those using immunosuppressive drugs.24
The intravenous infusion of immunoglobulins is performed every three to four weeks, with an initial dose of 400−600mg/kg, so that the IgG level is >500mg/dl in patients with agammaglobulinemia, with a consequent reduction in infections.25,26 Higher doses, of around 800mg/kg, help in the control of pulmonary problems, and are recommended for patients with chronic pulmonary disease and/or chronic sinusitis.2,27–29 The subcutaneous infusion of immunoglobulins can be carried out weekly, biweekly, or monthly, depending on the formulation. In Brazil formulations at 10%, 20%, or 10% linked to the previous infusion of hyaluronidase, are available. The dose regimen is similar and follows the equivalent of 100−150mg/kg per week.24
IgG level monitoring should be carried out at intervals of three months up to a maximum of six months, depending on the infectious picture. After the sixth infusion, a stable value is reached and the dose and interval must be adjusted to obtain the best clinical result.16,24
Most adverse effects associated with the intravenous infusion of immunoglobulins are related to the infusion rate. Patients who have never received this medication or those who are infected are at increased risk of adverse effects. These effects are partly related to the formation of antigen-antibody complexes and can be reduced if the patient is afebrile and undergoing treatment for the infection.16,24 Another risk factor is the frequent change of immunoglobulin brand, a common fact in Brazil.30 Adverse effects during the infusion mimic infectious conditions. Among the symptoms, tremors, arthralgia/myalgia, fever, and headache have been observed. Good patient hydration and reduced infusion rate are effective measures in preventing adverse events. Regarding the subcutaneous infusion, a fraction of the cases have local irritation effects, which tend to disappear over time.24
MedicationsIn the last decade, the great advance in the knowledge of genetics and the understanding of the pathophysiology of primary immunodeficiencies and of how changes in certain molecules generate alterations in important immune system pathways has also opened a window of opportunities for the more accurate treatment of these patients. This greater understanding of the immune system function allowed a deeper comprehension of changes in its regulation and the impact on the patients, who often did not fit in as a classic primary immunodeficiency or a classic autoimmune disease. Currently, these patients are included in a growing group of alterations known as immune dysregulation and thus, new treatment opportunities have also been found for these patients.1
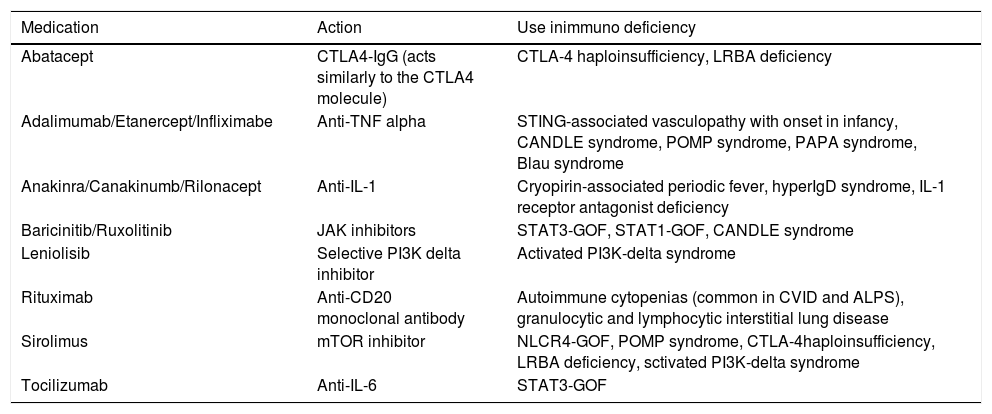
Therefore, new drugs appear all the time, especially precision immunobiological agents, which are being tested and approved for patients with IEI. Regarding the immunological knowledge, other drugs have been reassessed and some have been used for more specific defects of the immune system, such as several immunosuppressants in immune dysregulations. As they constitute a large number of diseases and drugs, extremely specific for one or other PID, the authors chose to describe only those of main use in Table 2.14
Examples of medications used to treat patients with primary immunodeficiencies.
| Medication | Action | Use inimmuno deficiency |
|---|---|---|
| Abatacept | CTLA4-IgG (acts similarly to the CTLA4 molecule) | CTLA-4 haploinsufficiency, LRBA deficiency |
| Adalimumab/Etanercept/Infliximabe | Anti-TNF alpha | STING-associated vasculopathy with onset in infancy, CANDLE syndrome, POMP syndrome, PAPA syndrome, Blau syndrome |
| Anakinra/Canakinumb/Rilonacept | Anti-IL-1 | Cryopirin-associated periodic fever, hyperIgD syndrome, IL-1 receptor antagonist deficiency |
| Baricinitib/Ruxolitinib | JAK inhibitors | STAT3-GOF, STAT1-GOF, CANDLE syndrome |
| Leniolisib | Selective PI3K delta inhibitor | Activated PI3K-delta syndrome |
| Rituximab | Anti-CD20 monoclonal antibody | Autoimmune cytopenias (common in CVID and ALPS), granulocytic and lymphocytic interstitial lung disease |
| Sirolimus | mTOR inhibitor | NLCR4-GOF, POMP syndrome, CTLA-4haploinsufficiency, LRBA deficiency, sctivated PI3K-delta syndrome |
| Tocilizumab | Anti-IL-6 | STAT3-GOF |
CTLA-4, cytotoxic T-lymphocyte–associated antigen 4; LRBA, LPS-responsive beige-like anchor protein; STING, stimulator of interferon genes; CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; POMP, proteasome maturation protein; PAPA, Pyogenic arthritis, pyoderma gangrenosum and acne; CVID, common variable immunodeficiency; ALPS, Autoimmune lymphoproliferative syndrome.
Adapted from Bundy et al.14
Most primary immunodeficiencies occur due to genetic defects intrinsic to hematopoietic cells; therefore, the replacement of these altered cells by hematopoietic stem cells from healthy donors, better known as bone marrow transplantation, is a very rational therapeutic approach.31 The first hematopoietic stem cell transplantations (HSCT) in patients with PID occurred more than 50 years ago, in patients with SCID and Wiskott-Aldrich syndrome.32,33 The approach to HSCT and the overall risk has changed substantially in the past two decades, with more potential donor sources, better targeting of preparatory chemotherapy regimens, and better support care.34,35
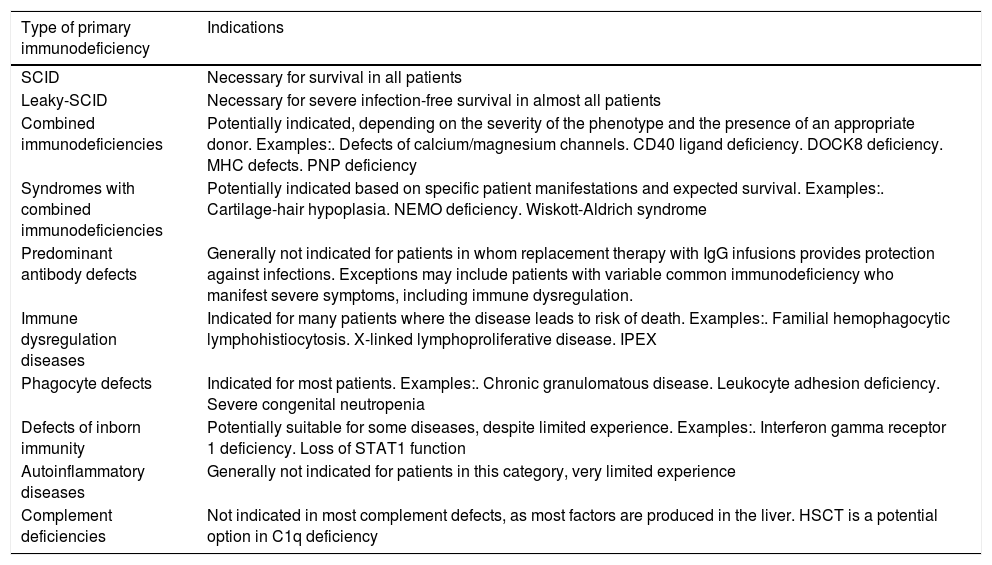
The decision about the indication and the correct timing of HSCT indication for a patient diagnosed with a primary immunodeficiency should always carefully consider the risks of HSCT against the risks of future evolution of the disease; it should be individualized and based not only on the specific PID, but also on each patient’s characteristics.31 In particular, patients with SCID represent a medical emergency, as they are highly susceptible to life-threatening infections; in these patients, HSCT provides a curative treatment.34,35Table 3 presents an overview of HSCT indications in different groups of primary immunodeficiencies.14
Indications for hematopoietic stem cell transplantation (HSCT) in primary immunodeficiencies.
| Type of primary immunodeficiency | Indications |
|---|---|
| SCID | Necessary for survival in all patients |
| Leaky-SCID | Necessary for severe infection-free survival in almost all patients |
| Combined immunodeficiencies | Potentially indicated, depending on the severity of the phenotype and the presence of an appropriate donor. Examples:. Defects of calcium/magnesium channels. CD40 ligand deficiency. DOCK8 deficiency. MHC defects. PNP deficiency |
| Syndromes with combined immunodeficiencies | Potentially indicated based on specific patient manifestations and expected survival. Examples:. Cartilage-hair hypoplasia. NEMO deficiency. Wiskott-Aldrich syndrome |
| Predominant antibody defects | Generally not indicated for patients in whom replacement therapy with IgG infusions provides protection against infections. Exceptions may include patients with variable common immunodeficiency who manifest severe symptoms, including immune dysregulation. |
| Immune dysregulation diseases | Indicated for many patients where the disease leads to risk of death. Examples:. Familial hemophagocytic lymphohistiocytosis. X-linked lymphoproliferative disease. IPEX |
| Phagocyte defects | Indicated for most patients. Examples:. Chronic granulomatous disease. Leukocyte adhesion deficiency. Severe congenital neutropenia |
| Defects of inborn immunity | Potentially suitable for some diseases, despite limited experience. Examples:. Interferon gamma receptor 1 deficiency. Loss of STAT1 function |
| Autoinflammatory diseases | Generally not indicated for patients in this category, very limited experience |
| Complement deficiencies | Not indicated in most complement defects, as most factors are produced in the liver. HSCT is a potential option in C1q deficiency |
SCID, severe combined immunodeficiency; DOCK8, dedicator of cytokinesis 8; MHC, major histocompatibility complex; NEMO, nuclear factor (NF)-kappa-B essential modifier; STAT1, signal transducer and activator of transcription 1; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome.
Gene therapy consists of the genetic modification of autologous hematopoietic stem cells of the individual with a vector containing the corrected gene product, a procedure performed in the laboratory and later administered to the patient as an autologous bone marrow transplant. The great advantage of this procedure is that there is no need to find compatible donors, reducing the time of search and the chance of graft versus host disease. However, the initial trials with retroviral vectors were complicated with cases of leukemia and myelodysplasia using retroviral vectors.36,37 Currently, the use of lentiviral vectors was shown to be safer, and trials with SCID due to ADA deficiency, X-linked SCID, WAS, and DCG have shown good immunological reconstitution in phase I, with no reported incidence of myelodysplasia/leukemia.38–40
ConclusionsThe expansion of knowledge on genetics and the pathophysiology of primary immunodeficiencies has increased the therapeutic arsenal for the treatment of these patients. Immunoglobulin replacement therapy remains the main therapeutic tool, as most patients with IEI have alterations in antibody quantity or quality. Precision medicine is already a reality for many patients with IEI in specific pathways, which can be treated with targeted medications for those pathways. Improvements in the management of HSCT in recent years have made it possible to transplant more and more patients with PID, offering curative therapy. In recent years, gene therapy has been successful and has become a hope for the future of patients with PIDs. Despite all the advances, it is important to remember that the diagnosis is the starting point for the treatment and should always be considered by pediatricians, who are responsible for the initial suspicion of primary immunodeficiencies.
FundingGRSS-Jeffrey Modell Foundation CHILDREN Program, ACN: Jeffrey Modell Foundation CHILDREN Program, CNPQ, FAPESP.
Conflict of interestThe authors declare no conflicts of interest.





