

To describe the hereditary angioedema to improve awareness of this condition and reduce diagnostic delay.
Data sourcesRelevant articles in the MEDLINE database through PubMed.
Data synthesisHereditary angioedema is rare and has an autosomal dominant pattern of inheritance. Its onset occurs mainly in childhood, but there is an important delay in the diagnosis. In the most frequent phenotype, there is a quantitative and/or functional deficiency in the C1esterase inhibitor protein, which regulates the activation of the complement, contact and fibrinolysis systems with greater formation of bradykinin, the main mediator of angioedema. There is a third type, the hereditary angioedema with a normal C1 inhibitor level, which is rare in children. Clinical manifestations are characterized by recurrent angioedema attacks, mainly in the extremities, abdomen and upper airways, which can progress to asphyxia and death. The main triggers are mechanical trauma, infections and stress. The diagnosis is attained by patient clinical picture and decreased serum levels of C4 and C1esterase inhibitor or its function. In hereditary angioedema with a normal C1 inhibitor, there is no change in these parameters, thus requiring a genetic study. Treatment is based on the use of attack medications and long and short-term prophylaxis.
ConclusionsHereditary angioedema is little known by pediatricians due to the significant delay in diagnosis of this condition, whose onset usually begins in childhood. The presence of recurrent angioedema that does not respond to treatment with antihistamines, corticosteroids and adrenaline should increase the diagnostic suspicion.
The first cases of hereditary angioedema were described by William Osler almost 140 years ago, but the diagnosis of this condition, which shows the first clinical manifestations in childhood, is still delayed.1,2 Therefore, many cases are not recognized by pediatricians, indicating the need for better knowledge of this disease.
In angioedema, there is a localized and transient increase in endothelial permeability, with fluid accumulation in the subcutaneous and submucosal tissues.3,4 It can occur concomitantly with papules, which constitute either acute or chronic urticaria if it lasts less than or more than six weeks, respectively.3 However, it is the onset of recurrent isolated angioedema that constitutes the main diagnostic challenge. In these cases, it is classified according to the main vasoactive mediator, that is, with or without histamine, and also regarding the presence of heredity.4 In the case of hereditary angioedema (HAE) the main mediator is bradykinin, and therefore there is no response to treatment with antihistamines or corticosteroids.1,2,5,6
HAE is a rare genetic condition with a variable frequency estimated at 1:50,000.1,2,5,6 It may be caused by a change in serum C1esterase inhibitor protein (C1-INH), which was initially described, as well as no change in this protein, defined some 20 years ago.2,5
In HAE with C1-INH deficiency (HAE-C1-INH) the autosomal dominant inheritance occurs due to a mutation in the SERPING1 gene, and 748 mutations have already been described where the majority of affected patients are heterozygous.7 This genetic heterogeneity partly explains the variability of clinical manifestations in these patients.
The frequency is unknown in HAE with normal C1-INH (HAE-n-C1-INH), but it also has an autosomal dominant pattern of inheritance.8 In this condition, mutations in protein genes involved in the formation or regulation of bradykinin action (factor XII, angiopoietin1, plasminogen, and kininogen) have already been identified, but no genetic changes have been found in most cases.8 HAE-nC1-INH can affect the pediatric population, but less frequently.9
PathophysiologyThe deficiency of C1-INH results in greater activation of the complement system, since this protein inhibits the initial activation complex of the first component of the complement in the classical pathway, hence the name C1-esterase inhibitor.1,2,5,6 Therefore, C1-INH regulates the system function, and it was subsequently described that it also acts in the control of the initial activation of other complement pathways, such as the alternative pathway and the lecithin pathway.5 In C1-INH deficiency, there is a non-controlled activation of the complement pathway, with C4 fraction consumption. However, bradykinin formation occurs mainly because C1-INH also controls the contact system activation.5,8
The contact system action begins with the activation of factor XII, which induces the generation of kallikrein from a serum precursor.5,8 In turn, kallikrein cleaves the high-molecular-weight kininogen, forming the bradykinin that acts on B2 receptors in endothelial cells, increasing vascular permeability.5,8 C1-INH regulates factor XII and kallikrein (Fig. 1).8

Sites of the C1 esterase inhibitor action in the complement system, contact system and fibrinolytic pathway.
C1-INH, C1 esterase inhibitor; HAE-C1-INH, hereditary angioedema due to C1-inhibitor deficiency; HMW kininogen, high-molecular-weight kininogen; C1qC1rCs, components q, r and s of the first fraction of the complement; C4, component C4 of the complement.
C1-INH physiologically inhibits the complement system, the fibrinolytic pathway and the contact system. The sites of C1-INH action in these pathways are indicated with a red line. In HAE-C1-INH there is a deficiency of C1-INH, with greater activation of these systems, which interact with each other with higher production of bradykinin that binds to B2 receptors in the endothelial cell, increasing vascular permeability with the formation of angioedema.
In addition to regulating the complement, contact and coagulation system pathways, C1-INH controls the fibrinolytic system, which consists of the activation of plasminogen with plasmin generation responsible for fibrin degradation.5,8 C1-INH controls the formation of plasmin by acting as an antifibrinolytic.
The contact, coagulation, complement and fibrinolysis systems interact among them (Fig. 1).5 Thus, an increased activation in these systems in HAE-C1-INH results in greater formation of bradykinin, in addition to serum reduction in the C4 fraction of the complement system.5,8
In HAE-n-C1-INH, an alteration in factor XII, plasminogen and kininogen results in increased bradykinin formation.8 Angiopoietin 1 acts as a negative regulator of bradykinin endothelial activation. Therefore, an alteration in angiopoietin 1 increases bradykinin action on the endothelial cell.8
Clinical manifestationsAs in adults, HAE-C1-INH in children manifests as recurrent episodes of subcutaneous and/or submucosal non-pruritic edema, lasting 2–5 days, with the skin, gastrointestinal tract and airways being the most commonly affected sites.10
Characteristically, HAE-C1-INH angioedema is not accompanied by urticaria, but around 60% of patients have macular, erythematous-serpiginous, fleeting and non-pruritic skin rashes known as erythema marginatum. Overall, they appear as a prodrome of angioedema attacks11; however, in younger children erythema marginatum is described as an independent phenomenon, without subsequent angioedema and as the inaugural manifestation of HAE-C1-INH.12
Subcutaneous edema in the extremities is the most common location in children. Recurrent, deforming, painless and non-pruritic, the angioedema has amil devolution and is often mistaken for allergy.1,13
When the edema affects the skin, it is painless; however, it is very painful in abdominal attacks, simulating acute abdomen and often resulting in unnecessary laparotomies.13,14 In these cases, imaging exams such as ultrasound or CT scan of the abdomen show the presence of ascites and intestinal wall edema, which, although unspecific for HAE-C1-INH, can be a particularly simple diagnostic tool in pediatric patients and useful to rule out acute surgical causes.15,16 Intestinal loop edema is the second most common site of HAE involvement in children, and, in addition to colic, abdominal distension, nausea, vomiting, watery diarrhea, prostration, and dehydration may occur, in some cases progressing to intestinal intussusception.1,13,17,18 Acute abdominal pain of unknown etiology is a frequent complaint in pediatric emergencies, and HAE-C1-INH should be among the diseases considered in the differential diagnosis.10
The involvement of the larynx in HAE-C1-INH rarely occurs in children; however, it can be the first disease presentation symptom and a life-threatening condition.13,19 Additionally, asphyxia can develop more rapidly than in adults, due to the smaller diameter of airways in children.10,20 Clinically, it does not differ from upper airway edema of inflammatory or allergic etiology; however, it is refractory to treatment with corticosteroids, antihistamines and adrenaline.
Other sites may be affected by edema in children, including genitals, urinary bladder, kidneys, muscles, joints, pericardium, pleura and central nervous system, albeit less frequently.10,20
Symptom onsetAlthough C1-INH deficiency is present since birth, the onset of HAE-C1-INH symptoms usually starts between 5 and 11 years of age.1,10,13,18,20 In a Brazilian sample of 95 pediatric patients with HAE-C1-INH I or II, 13.7% of the children had symptoms during the first year of life, and 53% between 1 and 5 years of age.21 Overall, in 50% of cases of HAE-C1-INH, the disease onset occurs at around 10 years of age, with an increase in the frequency and severity of attacks during puberty.22 Moreover, there is a direct association between early symptom onset and greater severity of the disease throughout life.1,10,22 HAE with normal C1-INH is rare in children, but most cases begin at adolescence.9 In a series of 197 Brazilian patients with HAE with normal C1-INH, 72% had their first attack between 12 and 25 years of age.23
Delayed diagnosisAlthough the onset of the first symptoms of HAE-C1-INH occurs in childhood or adolescence, the diagnosis is delayed, and most patients are diagnosed as adults.24
As this is a rare disease, unknown and often mistaken for allergy, the delay in diagnosis of HAE-C1-INH is common.20 Studies show that the mean time to the disease diagnosis after symptom onset is between 11 and 20 years.20,21,25 Another factor that leads to the delayed diagnosis or underdiagnosis is the absence of a family history of HAE-C1-INH, which occurs in 25% of cases, due to a de novo genetic mutation.24,25
Triggering factorsIn children, most attacks occur due to upper airway infections, mechanical trauma, and stress.18,20,21 In adolescents, menstruation and ovulation are frequent triggers of angioedema.26 Medications such as estrogen-containing oral contraceptives and angiotensin-converting enzyme (ACE) inhibitors are additional triggers.26,27
DiagnosisThe diagnosis of HAE-C1-INH should be guided by the abovementioned characteristic symptoms, family history (absent in 25% of cases) and biochemical tests.1,2,4,6,28
The diagnostic algorithm recommends the initial screening with serum or plasma levels of component C4 of the complement.1,2,4,6,28 If the level is below 50% of the reference values, the investigation should be followed by the evaluation of the C1-INH antigenic level, and later by the C1-INH functional activity, if necessary.1,2,4,6,28
Patients with HAE-C1-INH have decreased levels of C4, which is consumed due to the activation of the classic complement pathway without its physiological inhibitor, C1-INH5 (Table 1). C4 is an excellent screening test for patients with C1-INH deficiency, as almost 100% of patients with HAE-C1-INH I/II have low levels of this component, with sensitivity ranging from 81 to 96%.1,2,4,6,28,29
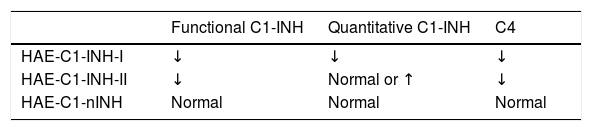
Laboratory diagnosis of HAE in children.
| Functional C1-INH | Quantitative C1-INH | C4 | |
|---|---|---|---|
| HAE-C1-INH-I | ↓ | ↓ | ↓ |
| HAE-C1-INH-II | ↓ | Normal or ↑ | ↓ |
| HAE-C1-nINH | Normal | Normal | Normal |
HAE-C1-INH-I, hereditary angioedema with C1 inhibitor type 1 deficiency; HAE-C1-INH-II, hereditary angioedema with C1 inhibitor type 2 deficiency; HAE-C1-nINH-I, hereditary angioedema with normal C1 inhibitor; Functional C1-INH, functional evaluation of the C1 inhibitor; Quantitative C1-INH, quantitative assessment of the C1 inhibitor; C4, serum level of component C4 of the complement.
In cases where C4 is normal, but the history of HAE is strongly suggestive, the test should be repeated during the angioedema attack, since C4 is invariably decreased in this situation.1,2,4,6,28 Then, the diagnosis must be confirmed through evidence of serum or plasma C1-INH levels below 50% of the reference values.1,2,4,6,28 Since C1-INH is an acute phase protein, blood collection for laboratory diagnosis should be performed in the absence of an inflammatory process.28
In HAE-C1-INH type I, which comprises around 85% of patients, both C1-INH serum levels and function are low (Table 1).1,2,4,6,28 In HAE-C1-INH type II, C1-INH levels are normal or elevated, and the diagnosis requires the assessment of C1-INHfunctional activity, which can be performed by immunoenzymatic or chromogenic assay, with the latter being more sensitive (Table 1).1,2,4,6,28
Overall, laboratory tests that analyze complement are very labile, especially the functional evaluation of C1-INH; therefore, it is recommended to ensure adequate conditions for the collection and handling of blood samples for more accurate results.1,2,4,6,28,30
The differentiation between HAE-C1-INH types I and II is merely diagnostic. The clinical presentation and treatment are the same for both phenotypes.
It is recommended that the diagnosis of HAE be based on two reduced and corresponding readings of C4 and antigenic and/or functional levels of C1-INH, collected at an interval of 1–3 months.1,2,4,6,28
Once the diagnosis is confirmed, active family screening for HAE-C1-INH is strongly recommended, due to the autosomal dominant type of inheritance, even in asymptomatic individuals, so that effective preventive or therapeutic measures can be implemented.1,2,4,6,28
The diagnosis of HAE-C1-INH in children is established in the same way as in adults, but there are some considerations.1,2,4,6,28 According to the ontogeny of children's immune systems, the complement system is immature during the first year of life, and therefore serum C4 and C1-INH levels, as well as functional C1-INH activity are physiologically low during childhood.1,31,32 Serum concentrations of C1-INH reach adult levels at between 6 and 12 months and C4 levels at around 2 and 3 years of age.33 For this reason, unlike what occurs in adults, C4 is not an accurate screening tool to diagnose HAE-C1-INH in children under 12 months of age.1,6 Additionally, most complement assays lack validated reference values for children.1,6
As in adults, the final diagnosis of HAE-C1-INH in children will only be established after two biochemical screening tests with corresponding results, the second of which should be performed only after 1 year of age.1,6 It is worth noting that every child under 1 year of age with a family history of HAE-C1-INH, even if asymptomatic, should be considered as having HAE-C1-INH until this diagnosis is ruled out.1,6
Despite the genetic nature of HAE-C1-INH, the disease is usually diagnosed based on clinical and biochemical findings.1,2,4,6,28 Overall, the molecular genetic analysis is not necessary to establish the diagnosis of HAE-C1-INH; however, it may be useful in cases where biochemical tests are inconclusive, such as in newborns and children under 1 year of age.1 If the family mutation is known, children can be diagnosed early with genetic tests performed on peripheral blood or cord blood.1,2,4,6,28,34 To date, this conduct has not been recommended as the first choice.1,2,4,6,28
Prenatal diagnosis in established pregnancies can be performed through genetic study on chorionic villus biopsy after the 10th week of pregnancy, as long as the family mutation is known.1,34,35 However, the risk of miscarriage is 0.5–1%.35 Moreover, the fact that the disease is treatable, not justifying the therapeutic interruption of the pregnancy, and the fact that therapeutic abortion in Brazil is not allowed for this condition make this diagnostic tool an unattractive one.
In the context of in vitro fertilization, preimplantation genetic diagnosis allows the selection of embryos without HAE-C1-INH, before implantation and pregnancy.35 However, this is an expensive procedure, it requires hormonal therapy for women, and the pregnancy rate is low.35
Mutations might not be identified in around 5–10% of molecular genetic tests, even in families that have been clearly established as havingHAE-C1-INH.7,8 The main method for detecting mutations in the SERPING1 gene is Classical Sanger Sequencing. However, if the mutation is not detected, more recent techniques such as next-generation sequencing (NGS) are required.34
Genetic testing is also of the utmost importance for the diagnosis of HAE-nC1-INH. In this case, other genes must be analyzed, such as F12, PLG, ANGPT1 and KNG1.34 The growing demand and dissemination of genetic tests for the diagnosis of several conditions have enabled a lower cost and the wider availability of these procedures in large centers. Once the genetic mutation is detected, genetic counseling is recommended to optimize extended family screening.34
TreatmentThe care of children with HAE requires the education of parents, teachers, caregivers and health professionals in relation to the disease to prevent severe consequences of HAE and improve the quality of life of the patients and their families. The parents/guardians must receive written information that is relevant to HAE, including preventive measures and an action plan for attack treatment. Older children can be trained to self-administer medications at home to ensure a rapid and effective treatment during attacks.2,6
The identification and the elimination of triggering factors reduce the risk of attacks. According to this line of recommendation, physical activities with a greater chance of mechanical trauma should be avoided, infections should be promptly treated, and the immunization of patients and those in contact with them is indicated, especially against influenza and hepatitis A and B viruses, as blood products can be used in the treatment of HAE.2 When mental stress is identified as a triggering factor, the need for psychotherapy or pharmacological treatment is recommended. A study including 33 children with HAE found a higher prevalence of anxiety when compared to healthy age-matched controls.36 Other studies have similarly demonstrated a significant negative impact on the quality of life of these patients.1 Drugs that increase bradykinin levels, inducing or prolonging an HAE attack, such as angiotensin I-converting enzyme (ACE) inhibitors, angiotensin II-receptor blockers (ARB), estrogen and gliptins should be avoided. Adolescents who need contraception should receive only progestogens.2
A better understanding of HAE pathogenesis in recent years has been essential for the development of specific therapies for the treatment of this disease, especially for acute attacks that cause great discomfort and are life-threatening for these patients. However, phase-3 studies about the use of these drugs in pediatric patients are limited.1 All evidence in the literature with controlled and randomized studies on HAE therapy refers to HAE-C1-INH.9 For HAE with no change in C1-INH, most case reports or case series are in adult patients.9 Drug treatment can be subdivided into attack treatment and short and long-term prophylactic treatment.1,2,6
Attack treatmentThe first step in the approach of patients undergoing an HAE attack is to assess the involvement of the airways, tongue, and/or uvula and thus guarantee a patent airway. In patients at imminent risk of suffocation, orotracheal intubation should not be delayed.19
The treatment of HAE has changed dramatically in recent years, with new and efficient drugs for attack management. There are 4 groups of drugs available for attack treatment: plasma-derived C1-inhibitor concentrate (pd C1-INH), recombinant C1 inhibitor (Rh C1-INH), bradykinin B2-receptor blocker (Icatibant), and kallikrein inhibitor (Ecallantide).6 However, few strategies have been approved for use in the pediatric population. Until recently, pd C1-INH was the only approved therapy for children.1 Icatibant was recently approved to be used in children aged 2 years and older.1 In Brazil, to date, there are two drugs approved by the National Health Surveillance Agency (ANVISA, Agência Nacional de Vigilância Sanitária): pd C1-INH (Berinert®) and Icatibant (Firazyr®).
The pd C1-INH for intravenous use is effective and safe in the treatment of all forms of HAE attacks due to C1-INH deficiency in children and adolescents.1 A recent study on the use of pd C1-INH in the pediatric age group for an extended period of time confirmed its effectiveness and safety.37 Berinert® is indicated for intravenous administration at a dose of 20UI/kg, regardless of the attack severity and without age restriction.1 Another nano-filtrated pdC1-INH, Cinryze®, has been approved for adolescents in some countries at fixed doses (500U or 1000U).1 However, there is evidence that fixed doses may not be sufficient to control attacks, with weight-based therapy (20U/kg) being the most effective one.6
Icatibant (Firazyr®) is a synthetic analogue of bradykinin and acts as a competitive and selective antagonist of the bradykinin B2 receptor. The safety and efficacy of Icatibant has been studied in children.38 Most patients started to show symptom resolution in approximately 1h, and the most common adverse event was a local reaction at the injection site with spontaneous resolution. The recommended dose is 0.4mg/kg in the age group of 2–17 years; with a body weight of more than 12kg, it is applied subcutaneously and exclusively in the abdominal region, and additional injections can be administered every 6h up to a maximum of three injections in a 24-h period.38 It is applied using 3mL syringes containing 10mg/mL of Icatibant. Doses can be adapted by weight [12–25kg=10mg (1mL); 26–40kg=15mg (1.5mL); 41–50kg=20mg (2mL); 51–65kg=25mg (2.5mL); >65kg=30mg (3mL)].38
As an alternative forC1-INH replacement therapy, there is the infusion of fresh frozen plasma at a dose of 10mL/kg. However, its effectiveness and safety have not been tested, and therefore this therapeutic option should be reserved for situations in which no other attack-specific drug is available.2 Moreover, plasma administration offers not only C1-INH replacement but also other substrates on which this protease acts, which can aggravate the condition, in addition to not having adequate efficacy.2
Long-term prophylaxisThe indication for long-term prophylactic treatment should be individualized, taking into account the frequency, severity and location of attacks, the possibility of patient access to emergency care, as well as the availability of effective therapies for the treatment of attacks.2 The goal of long-term prophylaxis is to decrease the frequency and severity of the attack.2 It should be noted that the number of events per year does not predict the severity of the next event or whether the next attack will affect the upper airways.2
The currently available drugs are attenuated androgens, antifibrinolytic agents, pdC1-INH and the monoclonal anti-kallikrein antibody, lanadelumab.1,39 The first two have been available for a long time, have lower costs and do not require intravenous access for their administration. However, there are concerns about their side effects and effectiveness.1,2,6
In Brazil, the most widely used attenuated androgen is danazol, which is available through the government's High Cost Drug Program.2 However, its long-term use in children is contraindicated due to adverse effects that are dose-dependent and include virilization and closing of the epiphyses, among others.1,2,6 Antifibrinolytic agents such as tranexamic acid have fewer side effects, but their effectiveness is not proven; however, they are safe options and are more frequently used in children with HAE with normal C1-INH.1,2,6,21
PdC1-INH has shown to be safe and effective for long-term prophylactic use in children and adolescents.1,37 The intravenous administration is performed at regular intervals (every 3–4 days), being considered the first-line medication.1,37 In 2017, the pdC1-INH for subcutaneous use, Haegarda®, was approved by the FDA (US Food and Drug Administration) for long-term prophylaxis, at a dose of 60U/kg every 2 weeks in adolescents.40 A recent study showed the efficacy and safety of the medication also in children.40 However, its use has not yet been approved in Brazil.
More recently, lanadelumab (Takhzyro®), which is a human anti-kallikrein monoclonal antibody for long-term prophylaxis, was approved in Brazil for children aged 12 years and older.39 The recommended starting dose is 300mg every 2 weeks, subcutaneously. In patients who have achieved attack control with treatment a reduction to 300mg every 4 weeks can be considered. Local reactions were the most common adverse effects.39
PdC1-INH (Haegarda®) and lanadelumab (Takhzyro®) have significantly changed long-term prophylaxis, since both are safe and the route of administration is subcutaneous.39,40 However, additional studies are still needed to evaluate the effectiveness and safety in younger children. Clinical studies are being carried out with oral medications, which could further change the treatment scenario for these patients.1
Short-term prophylaxisIt is indicated for patients who will undergo medical or surgical procedures that involve mainly the cervicofacial region, with risk of laryngeal edema.1,2,6 Examples of such procedures include dental manipulation, tonsillectomy, upper digestive endoscopy and surgical procedures that require orotracheal intubation.2 The decision on short-term prophylaxis must be made taking into account two factors: the risk associated with the procedure to be performed and the availability of treatment for the acute attack.2
Intravenous pd C1-INH is safe and effective in children and adolescents and it is the agent of choice for short-term prophylaxis.1,2,6 Its application is indicated 1–2h before the procedure at a dose of 20U/kg or 500–1000U, depending on the manufacturer. When not available, fresh frozen plasma has been used instead.2 However, it should be noted that this strategy has not been tested in clinical trials regarding the efficacy and safety in HAE.2 It can be administered at a dose of 10mL/kg approximately 1–6h before the procedure.2
Among the antifibrinolytic agents, tranexamic acid has been used at a dose of 25mg/kg/day divided into 2–3 times, 5 days before and 2–5 days after the procedure.2 However, its effectiveness is limited. Androgens, although more effective, should be used with caution in the pediatric population due to their side effects. The danazol dose is 10mg/kg/day, 5–7 days before and 3–5 days after the procedure.2
Even with prophylactic therapy, a dose of the medication for attack treatment should be available, since the risk of angioedema after these procedures cannot be completely eliminated.6
Final considerationsIn recent years, there has been a marked progress in the knowledge of the genetics and pathophysiology of HAE, which has allowed more effective and safer treatments. However, even though the disease onset starts in childhood, HAE remains an unknown disease, which is underdiagnosed by pediatricians. The aim of this review was to raise awareness of HAE among these professionals to promote greater diagnostic suspicion in children and thus improve the quality of life for this population.
Conflicts of interestRégis A. Campos: lecturer at Takeda; Solange O.R. Valle: lecturer at Takeda, CSL Behring; Eliana Toledo: lecturer at Takeda.




