


To describe the demographic, clinical, laboratory and molecular characteristics of patients with lysosomal acid lipase deficiency.
MethodsA retrospective review of the medical records of children with the disease.
ResultsSeven children with lysosomal acid lipase deficiency (5 male; 2 female); 6 were mixed race, and 1 was black. The mean ages at the first onset of symptoms and at diagnosis were 5.0 years (4 months to 9 years) and 6.9 years (3–10 years), respectively. Symptom manifestations at onset were: 3 patients had abdominal pain, one had bone/joint pain due to rickets, and 1 had chronic diarrhea and respiratory insufficiency due to interstitial pneumonitis. One was asymptomatic, and clinical suspicion arose due to hepatomegaly. Six patients had hepatomegaly, and none had splenomegaly. Two patients were siblings. Enzymatic assay and molecular analysis confirmed the diagnoses. Genetic analysis revealed a rare pathogenic variant (p.L89P) in three patients, described only once in medical literature and never described in Brazil. None of those patients were related to each other. Lysosomal acid lipase deficiency was previously described as an autosomal recessive disease, but three patients were heterozygous and undoubtedly had the disease (low enzyme activity, suggestive lab findings and clinical symptoms).
ConclusionThis case series supports that lysosomal acid lipase deficiency can present with highly heterogeneous signs and symptoms among patients, but it should be considered in children presenting with gastrointestinal symptoms associated with dyslipidemia. We describe a rare variant in three non-related patients that may suggest a Brazilian genotype for lysosomal acid lipase deficiency.
Descrever as características demográficas, clínicas, laboratoriais e moleculares de pacientes com deficiência de lipase ácida lisossomal.
MétodosAnálise retrospectiva dos prontuários médicos de crianças com a deficiência de lipase ácida lisossomal.
ResultadosSete crianças com deficiência de lipase ácida lisossomal (5M:2F); seis eram pardas e uma negra. As faixas etárias no início dos sintomas e no diagnóstico foram 5 anos (4 meses a 9 anos) e 6,9 anos (3 a 10 anos), respectivamente. As manifestações dos sintomas no início foram as que seguem: três pacientes apresentaram dor abdominal, um apresentou dor nos ossos/articulações devido a raquitismo e um apresentou diarreia crônica e insuficiência respiratória devido à pneumonite intersticial. Os outros não apresentaram sintomas e a suspeita clínica surgiu devido à hepatomegalia. Seis pacientes apresentaram hepatomegalia e um apresentou esplenomegalia. Dois pacientes eram irmãos. O ensaio enzimético e a análise molecular confirmaram os diagnósticos. A análise genética revelou uma variante patogênica rara (p.L89P) em três pacientes, descrita uma única vez na literatura médica e nunca descrita no Brasil. Nenhum desses pacientes tinha parentesco com os outros. A deficiência de lipase ácida lisossomal foi anteriormente descrita como uma doença recessiva autossômica, porém três pacientes eram heterozigotos e, sem dúvida, apresentaram a doença (atividade enzimática baixa, achados laboratoriais sugestivos e sintomas clínicos).
ConclusãoEsta casuística afirma que a deficiência de lipase ácida lisossomal pode se manifestar com sinais e sintomas altamente heterogêneos entre os pacientes, porém deve ser considerada em crianças que apresentam sintomas gastrointestinais associados à dislipidemia. Descrevemos uma variante rara em três pacientes não relacionados que pode sugerir um genótipo brasileiro para deficiência de lipase ácida lisossomal.
Lysosomal acid lipase deficiency (LALD) is an autosomal recessive disease belonging to the lysosomal storage diseases group.1–3 The phenotype of LALD varies according to the lysosomal acid lipase (LAL) enzyme activity, and the clinical spectrum can range from Wolman disease (WD), a severe infantile-onset form, to cholesteryl ester storage disease (CESD), a disease with milder symptoms but sometimes with severe presentation and bad prognosis.1–6 The low frequency of LALD has been challenged as population studies propose that its incidence may be higher than previously described.2,3,7 Additionally, the increase in clinical awareness has resulted in more diagnosis, supporting the idea that it may be an underdiagnosed disease. Many variants in the lysosomal acid lipase gene (LIPA) have been described, and new studies are still describing new mutations; currently, a genotype-phenotype correlation has not been found.1,2,8,9
Herein, we analyze 7 patients with LALD, which we believe is the largest series reported in South America.10
MethodsWe performed a retrospective review of the medical records of patients with LALD being cared for in our outpatient clinic. All data regarding demographic, clinical, laboratory, and molecular characteristics were retrieved and then analyzed. All patients and their parents (or guardians) were aware of the intent to publish their records, agreed to it and signed an informed consent form, according to the guidelines of the Ethical Committee of our institution, which approved this study.
The Seattle Children's Department of Laboratories conducted all the LAL activity tests and molecular analysis using dried blood spot (DBS) samples. LAL activity tests were performed using the method described by Hamilton et al. in 2012: 4-methylumbelliferyl palmitate was used as a substrate to determine total lipase activity; since whole blood has other lipases, Lalistat 2, an inhibitor of LAL, was added to the sample to measure pancreatic lipase activity, and finally, LAL activity was calculated. The units used were pmole/hr/spt. Sanger sequencing of the LIPA gene was carried out for every patient with low LAL activity.11
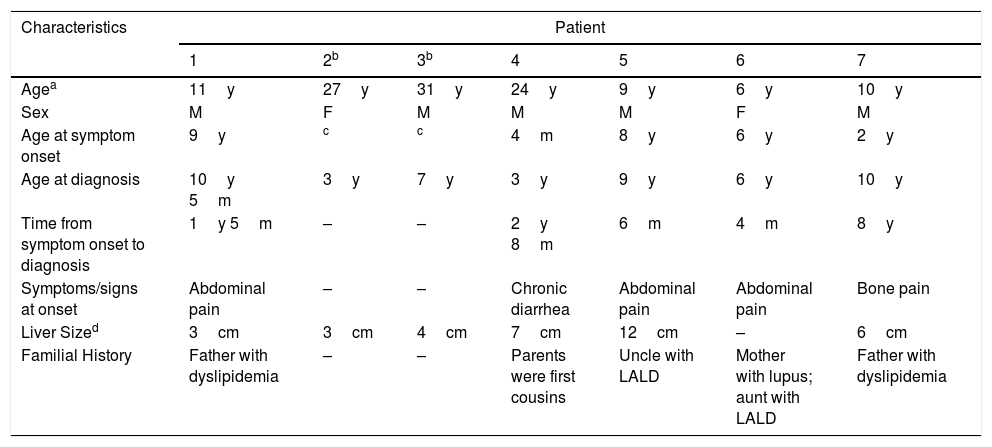
Results (case description)We reviewed the medical records of 7 children with LALD (5 male; 2 female); 6 were mixed race, and 1 was black. The mean and median ages were 5.0 years (4 months to 9 years) and 6 years at the first onset of symptoms and 6.9 years (3–10 years) and 6.5 years at diagnosis, respectively. The time between diagnosis and symptom onset varied widely and is shown in Table 1, along with other clinical features. Two patients were asymptomatic, and clinical suspicion arose due to hepatomegaly. Hepatomegaly was present in 6 patients, and none had splenomegaly or developed jaundice. Consanguinity was present in 2 patients who were siblings. Five out of 7 patients had family history for dyslipidemia. After clinical and laboratory suspicion in our patients, LAL activity was measured to confirm LALD.
Clinical characteristics.
| Characteristics | Patient | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2b | 3b | 4 | 5 | 6 | 7 | |
| Agea | 11y | 27y | 31y | 24y | 9y | 6y | 10y |
| Sex | M | F | M | M | M | F | M |
| Age at symptom onset | 9y | c | c | 4m | 8y | 6y | 2y |
| Age at diagnosis | 10y 5m | 3y | 7y | 3y | 9y | 6y | 10y |
| Time from symptom onset to diagnosis | 1y 5m | – | – | 2y 8m | 6m | 4m | 8y |
| Symptoms/signs at onset | Abdominal pain | – | – | Chronic diarrhea | Abdominal pain | Abdominal pain | Bone pain |
| Liver Sized | 3cm | 3cm | 4cm | 7cm | 12cm | – | 6cm |
| Familial History | Father with dyslipidemia | – | – | Parents were first cousins | Uncle with LALD | Mother with lupus; aunt with LALD | Father with dyslipidemia |
LALD, lysosomal acid lipase deficiency.
A hallmark of LALD is the clinical heterogeneity of symptoms at presentation. A detailed clinical description of the cases is mandatory.
Patient 1 came to our outpatient facility with abdominal pain, without any alarming signs, and was 8 years old. After continuous evaluation, hepatomegaly was noted, and an upper endoscopy showed sea-blue histiocytes in mucosal biopsy histological examination of the duodenum. LAL activity measurement confirmed LALD.
Patient 3, 6 years old; hepatomegaly was noted during routine pediatric consultation. Initial exams showed a 2–3-fold increase in alanine aminotransferase (ALT), low high-density lipoprotein (HDL), and high low-density lipoprotein (LDL). Liver biopsy showed lipid deposits in histiocytes. Later, LAL activity measured in the same patient when he was 7 years old was diagnostic for LALD. His sister, patient 2, also had slightly elevated liver enzymes at the age of 3 years, without hepatomegaly, and was diagnosed with LALD at the same time as her brother.
Patient 4, who was 4 months old, presented with progressive and intractable diarrhea (first occurring in 1991), failure to thrive, hepatomegaly, dyslipidemia and adrenal calcifications. When he was 3 years old, a liver biopsy revealed significant microvesicular steatosis, septal fibrosis, and aggregates of macrophages with cytoplasmic microvacuolation – findings suggestive of Niemann-Pick B at that time, as he also had low sphingomyelinase and high acid phosphatase levels. However, electron microscopy showed negative images of cholesterol crystals and lipid vacuoles in hepatocytes and histiocytes; thus, LALD was favored by this method. His chronic diarrhea and progressive liver disease continued. When he reached the age of 6, he developed numerous episodes of wheezing. Chest X-ray and tomography showed signs of interstitial pneumonitis. Spirometry revealed a restrictive pattern. When he was 13 years old, he developed complications due to liver failure with ascites and had spontaneous bacterial peritonitis twice. A liver from a deceased donor was transplanted when he was 18 years old at the beginning of 2010. His explanted liver (Fig. 1A and B) had micronodular cirrhosis and characteristic features of the disease, including some needle-shaped clefts inside microvacuolated Kupffer cells. Prior to liver transplantation, the patient developed small splenomegaly (2cm from the left costal edge) which decreased around 50% after liver transplantation. After the liver transplant, he had several rejection episodes, which were improved with corticosteroids. His liver biopsy in 2015 showed chronic rejection with ductopenia (50% loss), portal and centrilobular fibrosis, rare septa, central perivenulitis and a few foamy Kupffer cells (which are usually a response to cholestasis in this context). However, his liver biopsy in 2017 demonstrated many xanthomatous macrophages within the portal tract, strongly suggesting the recrudescence of the disease (Fig. 1C), along with marked bridging fibrosis (F3), chronic rejection and rare foamy sinusoidal cells. LAL enzyme activity measured in 2015 was zero. The patient's lipid profile improved to normal LDL and triglycerides (TG) levels after liver transplantation, but low HDL levels remained. Additionally, a duodenal biopsy performed in 2016 showed countless xanthomatous macrophages in the lamina propria (Fig. 1D).
 An advanced micronodular cirrhosis observed in the liver explant (H&E, 25×); (B) This field (of the liver explant) depicts microvesicular and mediovesicular steatosis in many hepatocytes and groups of enlarged xanthomatous Kupffer cells in sinusoids (H&E, 200×), in addition to rare needle-shaped clefts (arrows) inside Kupffer cells in the inset; (C) Many portal tract macrophages exhibit xanthomatous cytoplasm in the liver graft after 7 years of liver transplantation (H&E, 400×); (D) The lamina propria is intensely infiltrated by large lipid-laden foam cells in this duodenal biopsy (H&E, 400×).")
Some histological aspects of liver and duodenum in patient 4. (A) An advanced micronodular cirrhosis observed in the liver explant (H&E, 25×); (B) This field (of the liver explant) depicts microvesicular and mediovesicular steatosis in many hepatocytes and groups of enlarged xanthomatous Kupffer cells in sinusoids (H&E, 200×), in addition to rare needle-shaped clefts (arrows) inside Kupffer cells in the inset; (C) Many portal tract macrophages exhibit xanthomatous cytoplasm in the liver graft after 7 years of liver transplantation (H&E, 400×); (D) The lamina propria is intensely infiltrated by large lipid-laden foam cells in this duodenal biopsy (H&E, 400×).
Patient 5, 8 years old, presented with chronic abdominal pain along with hepatomegaly and increased aspartate aminotransferase (AST) and ALT, along with low HDL and high LDL. After an inconclusive initial investigation, a liver biopsy revealed findings suggestive of Niemann-Pick disease. As the clinical course was not suggestive of Niemann-Pick, a LAL activity measure diagnosed LALD. His aunt also had a prior diagnosis of Niemann-Pick disease that was later changed to LALD.
Patient 6 presented with chronic abdominal pain at around the age of 6 years. Initial lab evaluation showed slightly increased AST and ALT, low HDL and high LDL, and steatosis in liver ultrasound. He was not overweight and did not have an increased consumption of fructose, which would have been suggestive signs of non-alcoholic liver disease (NAFLD) or liver steatosis. Therefore, we searched LAL activity and confirmed LALD.
Patient 7, a 2-year-old boy, was admitted to our hospital due to difficulty in walking. Upon initial physical examination, hepatomegaly was noticed. Laboratory analysis clearly indicated rickets. When the patient was 3 years old, he was transferred to our hepatology clinic to investigate hepatomegaly. A liver biopsy performed when the patient was 6 years old suggested metabolic diseases, especially glycogenosis. However, the patient never developed hypoglycemia. In 2016, after measuring LAL activity, we diagnosed LALD. After a while, the pathologist re-evaluated the slides of the liver biopsy. The histopathological findings (Fig. 2) of scarce foamy Kupffer cells and some portal tract macrophages that are faintly positive for periodic acid-Schiff after diastase digestion (PASD), the absence of the characteristic swollen liver cells resembling plant cells, not to mention the presence of grade 3 steatosis are in favor of the diagnosis of LALD (in addition to other differential diagnoses, e.g. mucopolysaccharidoses). It is worth mentioning that this case only exhibited subtle changes in Kupffer cells, making the diagnosis more challenging, maybe related to sampling variability of liver biopsies.
 Marked bridging fibrosis with occasional nodules (Picrosirius red stain, 25×); (B) Mediovesicular steatosis (a small-droplet variant of the macrovesicular steatosis) and hepatocytes with pale cytoplasm (H&E, 400×); (C) Only rare microvacuolated Kupffer cells identified, which were slightly stained with PASD (PAS after diastase digestion, 400×); (D) Some macrophages with foamy, tan-colored cytoplasm were detected in portal tracts (H&E, 400×).")
Histological aspects of the liver biopsy in patient 7. (A) Marked bridging fibrosis with occasional nodules (Picrosirius red stain, 25×); (B) Mediovesicular steatosis (a small-droplet variant of the macrovesicular steatosis) and hepatocytes with pale cytoplasm (H&E, 400×); (C) Only rare microvacuolated Kupffer cells identified, which were slightly stained with PASD (PAS after diastase digestion, 400×); (D) Some macrophages with foamy, tan-colored cytoplasm were detected in portal tracts (H&E, 400×).
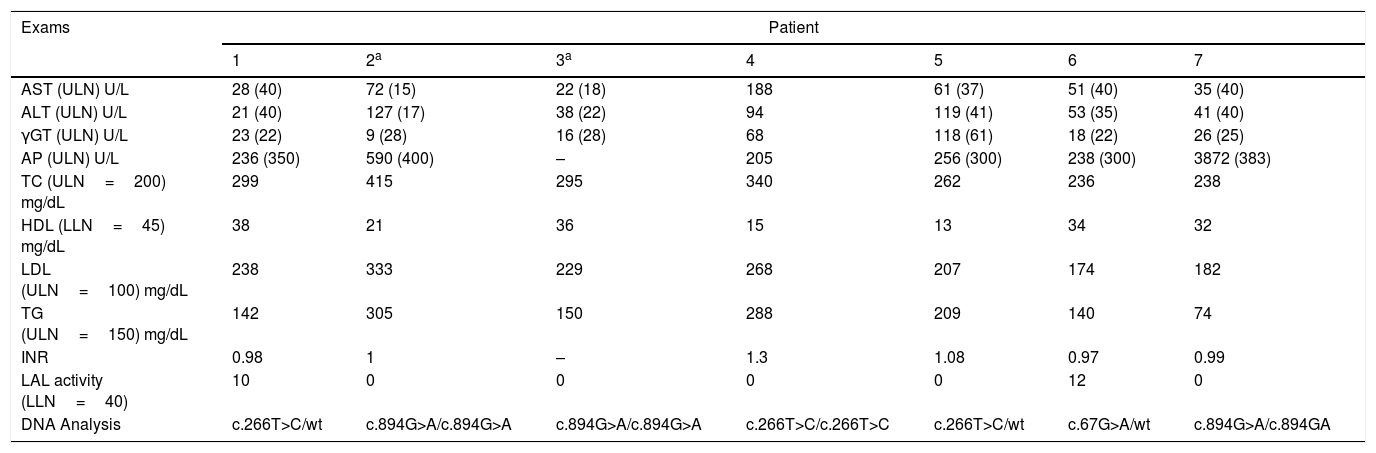
Table 2 summarizes the laboratory features of our patients. Genetic analysis revealed one rare variant p.L89P (c.266T>C) in 3 patients, which had never been described in Brazil.12 None of those 3 patients were related to each other. One patient was homozygous for p.L89P, and the others were heterozygous. The variants c.894G>A, a splice site variant at the 3’ end of exon 8, and c.67G>A found in our patients have already been commonly described.
Laboratory workup at symptom onset and genetic analysis.
| Exams | Patient | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2a | 3a | 4 | 5 | 6 | 7 | |
| AST (ULN) U/L | 28 (40) | 72 (15) | 22 (18) | 188 | 61 (37) | 51 (40) | 35 (40) |
| ALT (ULN) U/L | 21 (40) | 127 (17) | 38 (22) | 94 | 119 (41) | 53 (35) | 41 (40) |
| γGT (ULN) U/L | 23 (22) | 9 (28) | 16 (28) | 68 | 118 (61) | 18 (22) | 26 (25) |
| AP (ULN) U/L | 236 (350) | 590 (400) | – | 205 | 256 (300) | 238 (300) | 3872 (383) |
| TC (ULN=200) mg/dL | 299 | 415 | 295 | 340 | 262 | 236 | 238 |
| HDL (LLN=45) mg/dL | 38 | 21 | 36 | 15 | 13 | 34 | 32 |
| LDL (ULN=100) mg/dL | 238 | 333 | 229 | 268 | 207 | 174 | 182 |
| TG (ULN=150) mg/dL | 142 | 305 | 150 | 288 | 209 | 140 | 74 |
| INR | 0.98 | 1 | – | 1.3 | 1.08 | 0.97 | 0.99 |
| LAL activity (LLN=40) | 10 | 0 | 0 | 0 | 0 | 12 | 0 |
| DNA Analysis | c.266T>C/wt | c.894G>A/c.894G>A | c.894G>A/c.894G>A | c.266T>C/c.266T>C | c.266T>C/wt | c.67G>A/wt | c.894G>A/c.894GA |
AST, aspartate aminotransferase; ALT, alanine aminotransferase; γGT, gamma-glutamyl-transferase; AP, alkaline phosphatase; TC, total cholesterol; HDL, high density lipoprotein; LDL, low density lipoprotein; TG, triglycerides; INR, international normalized ratio; ULN, upper limit normal; LLN, lower limit normal; wt, wild type gene.
We report 7 patients with LALD, the largest case series ever described in South America. Patients’ diagnoses were confirmed after high clinical suspicion that led to the measurement of LAL activity, which was invariably low. Our clinical awareness of LALD has improved throughout the years, and this explains the variable time range between symptom onset and diagnosis. The 3 patients for whom it took more than 1 year to reach diagnosis are the ones who had atypical symptoms at onset.
Patient 4 had severe symptoms that resembled Wolman disease, but he also had interstitial pneumonitis, low sphingomyelinase, and high acid phosphatase levels, which are confounding factors because those findings are suggestive of Niemann-Pick disease.
Patient 7 had hepatomegaly, and the initial liver biopsy examination suggested a storage disease such as glycogenosis since some findings (e.g. the altered Kupffer cells) were not very conspicuous in the tissue sample. The re-evaluation of the liver slides, after consideration of the new information of LALD, was compatible with the disease.
Those 2 cases mentioned above (patients 4 and 7) add LALD as a potential differential diagnosis of glycogenosis and Niemann-Pick type B. Gaucher disease, Niemann-Pick type C and NAFLD have already been described as differential diagnoses of LALD.13,14 Differential diagnoses of LALD are broad and usually overlap with other chronic conditions. Consequently, one reason why the real prevalence of LALD is unknown is because it is an underdiagnosed disease.2,3,7–9,15,16 There must be high clinical suspicion of LALD in patients with unexplained hepatomegaly, high LDL and triglyceride levels, low HDL levels and mildly elevated liver enzymes.1,2,8 Six of our patients had hepatomegaly; all had a typical cholesterol pattern, and five had mildly elevated liver enzymes. These results show that there must be a pattern of findings for diagnosing LALD, even in atypical cases.
LIPA, the LAL encoding gene, is located on chromosome 10q23.31. Many variants in this gene are known to be associated with LALD. The c.894+1G>A (E8SJM+1) variant is the most common and is responsible for more than half of the cases published; 3 of our patients had this variant.1,2 The other variant, c.67G>A, has already been reported three times in the ClinVar website.17 Interestingly, we herein describe a rare variant, c.266T>C (p.L89P), which was found in 3 patients. This pathogenic variant was described only once in a poster presentation at the American Society of Human Genetics Meeting in 2014.12 We propose a possible Brazilian genotype for LALD, as none of the patients with this variant were related to each other. The patient who was homozygous for the latter variant had the most severe presentation, with pulmonary involvement, chronic diarrhea and progressive liver disease, which required a liver transplantation (patient 4).
LALD is classically defined as an autosomal recessive disease. However, patients 1, 5 and 6 had one mutant allele and one wild-type allele. All three of those patients had laboratory and clinical signs suggestive of LALD (hepatomegaly, low HDL, high LDL, and low LAL activity), and other probable diseases were excluded. Niemann-Pick disease was a major confounding diagnosis for patient 5, as he had an aunt who had Niemann-Pick disease. However, after our diagnosis, we investigated his aunt and were able to diagnose her with LALD. NAFLD and fructose intolerance were other excluded diagnosis as they can appear with hepatomegaly and liver steatosis, and specifically NAFLD can appear with dyslipidemia. One hypothesis is that those patients may have intronic mutations that were not recognized through Sanger sequencing, or the reduction of LAL activity in some heterozygous patients may lead to symptoms. Pullinger et al. already described patients heterozygous for LIPA gene pathogenic variants that had symptoms and lab data suggestive of LALD.2,18
The treatment and prognosis of LALD has dramatically changed in the last years after the creation of sebelipase alfa (Kanuma®, Kanuma™), an enzyme replacement therapy (ERT) that has yielded improvements of liver damage and lipid abnormalities.19–22 Liver transplantation for LALD is another possible treatment for severe cases and has been reported only 20 times in the medical literature, but few papers report long-term follow-up for those patients.2 Bernstein et al. published 2 cases of LALD patients submitted to liver transplantation, and in both of them multisystemic LALD disease recurred after transplantation. They also reviewed the literature of 18 cases of LALD patients submitted to liver transplantation and described multisystemic LALD progression in 11 patients (66%) and death in 6 patients (33%).23 In our study, patient 4 underwent liver transplantation, and after 5 years of follow-up, he had chronic rejection and maintained low HDL levels. Interestingly, this patient still had cholesterol deposition in macrophages and showed xanthomatous histiocytes in a duodenum biopsy in 2016, and later developed foamy portal tract macrophages in the liver biopsy after 7 years since the liver transplant. Therefore, a liver transplant does not cure the disease, as macrophages continue to be impaired, leading to symptoms since other organs, e.g. bone marrow and small intestine, will still contain the lipid-laden macrophages. On the other hand, bone marrow transplantation (BMT) is believed to correct the enzyme defect and therefore may preserve the life of patients with Wolman disease if performed early in the disease course. Some successful reports of BMT in LALD have been reported; however, the majority of the cases have a bad prognosis and prompt ERT is the therapy of choice.24–26
In conclusion, we report the largest case series of patients with LALD in South America and describe a rare genotype, possibly a Brazilian genotype. We emphasize that LALD can present with heterogeneous signs and symptoms but should be included as a differential diagnosis of hepatomegaly, elevated liver enzymes, and dyslipidemia.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Benevides GN, Miura IK, Person NC, Pugliese RP, Danesi VL, Lima FR, et al. Lysosomal acid lipase deficiency in Brazilian children: a case series. J Pediatr (Rio J). 2019;95:552–8.





