

To verify genetic determinants associated with stroke in children with sickle cell disease (SCD).
MethodsProspective cohort with 110 children submitted to neonatal screening by the Neonatal Screening Program, between 1998 and 2007, with SCD diagnosis, followed at a regional reference public service for hemoglobinopathies. The analyzed variables were type of hemoglobinopathy, gender, coexistence with alpha thalassemia (α-thal), haplotypes of the beta globin chain cluster, and stroke. The final analysis was conducted with 66 children with sickle cell anemia (SCA), using the chi-squared test in the program SPSS® version 14.0.
ResultsAmong children with SCD, 60% had SCA. The prevalence of coexistence with α-thal was 30.3% and the Bantu haplotype (CAR) was identified in 89.2%. The incidence of stroke was significantly higher in those with SCA (27.3% vs. 2.3%; p=0.001) and males (24.1% vs. 9.6%; p=0.044). The presence of α-thal (p=0.196), the CAR haplotype (p=0.543), and socioeconomic factors were not statistically significant in association with the occurrence of stroke.
ConclusionThere is a high incidence of stroke in male children and in children with SCA. Coexistence with α-thal and haplotypes of the beta globin chain cluster did not show any significant association with stroke. The heterogeneity between previously evaluated populations, the non-reproducibility between studies, and the need to identify factors associated with stroke in patients with SCA indicate the necessity of conducting further research to demonstrate the relevance of genetic factors in stroke related to SCD.
Verificar fatores genéticos associados ao acidente vascular encefálico (AVE) em crianças com Doença Falciforme (DF).
MétodosCoorte prospectiva de 110 crianças submetidas à triagem neonatal pelo Programa de Triagem Neonatal, entre 1998-2007 com o diagnóstico de DF, atendidas em serviço público regional de referência em hemoglobinopatias. As variáveis analisadas foram: tipo de hemoglobinopatia, sexo, coexistência da alfa Talassemia (α-Tal), haplótipos do cluster da cadeia beta globina e AVE. A análise estatística final foi realizada com 66 crianças com Anemia Falciforme, por meio do teste do Qui-quadrado no programa SPSS® 14.0.
ResultadosEntre as crianças com DF, 60% eram portadoras de Anemia Falciforme. A prevalência da coexistência com a α-Tal foi de 30,3% e o haplótipo Bantu (CAR) foi identificado em 89,2%. A incidência de AVE foi significativamente maior nas crianças com AF (27,3% versus 2,3%; p = 0,001) e no sexo masculino (24,1% versus 9,6%; p = 0,044). A presença da α-Tal (p = 0,196), do haplótipo CAR (p = 0,543) e fatores socioeconômicos não foram significantemente associadas à ocorrência de AVE.
ConclusãoO AVE apresenta alta incidência em crianças com AF e em crianças do sexo masculino. Coexistência de α-Tal ou de haplótipos do cluster da beta globina não apresentaram associação significante com AVE. A heterogeneticidade entre as populações previamente avaliadas e a não reprodutibilidade entre estudos indicam a necessidade de realização de novas pesquisas para verificar o papel desses fatores genéticos no AVE em crianças com DF.
Sickle cell disease (SCD) is the most common monogenic hereditary disease in Brazil, occurring predominantly among those of African descent. The term SCD includes sickle cell anemia (SCA) and pathological conditions in which the hemoglobin S gene is associated with other hemoglobinopathies, such as SC, S/beta0 and S/beta+ thalassemia (S/b), and SD Punjab, among others.1 SCA, caused by a single mutation in the β-globin gene, produces a diversity of phenotypic expressions in affected patients.2,3 SCA is the most severe form of SCD presentation; for the disease to manifest, homozygosity of βS alleles in the gene responsible for the synthesis of β chain of hemoglobin is required, determining the formation of hemoglobin S (HbSS).
HbSS, under conditions such as low oxygenation, metabolic acidosis, or dehydration, becomes polymerized, irreversibly changing the structure of erythrocyte, thus determining inefficient oxygenation, endothelial inflammatory reaction, and the entire complex physiopathology of the disease.3,4 The polymerization process leads to vascular occlusion, which can trigger painful crises, stroke, acute chest syndrome, splenic sequestration, and priapism, among other manifestations.
In Brazil, studies have shown that 700–1000 children/year are born with SCD,1,4–6 making this disease a public health problem. The state of Minas Gerais (MG) is a pioneer in Brazil in the early diagnosis of SCD, with the introduction of the Neonatal Screening Program of the State of Minas Gerais (Programa de Triagem Neonatal do Estado de Minas Gerais [PETN-MG]) in March 1998. PETN-MG is coordinated by the Center for Action and Research in Support Diagnostics (Núcleo de Ações e Pesquisa em Apoio Diagnóstico [NUPAD]) of Universidade Federal de Minas Gerais, which refers newborns diagnosed with SCD to be followed at Fundação Hemominas. From 1998 to 2007, 2,549,097 children were screened in MG, of whom 188,916 were born in the 39 municipalities included in the regional reference public services for hemoglobinopathies – where this study was carried out – which represents 7.41% of the total number in the state. The PETN-MG coverage in 2007 was 88.57%, which is higher than the national average of 9.65% (Ministry of Health, National Neonatal Screening Program, 2009).
Stroke is one of the most severe complications of SCD and is responsible for 20% of mortality among these patients.2,7 According to the Cooperative Study Group in SCD, the overall incidence of the first stroke was 0.08 acute events/100 patients/year in children under 2 years; 0.75 in patients between 2 and 5 years of age; 0.55 between 6 and 9 years of age; 0.30 between 10 and 19 years; and 0.45 between 20 and 29 years. Among the SCD, the incidence of stroke is 0.61 for patients with Hb SS, 0.17 for Hb SC, and 0.11 for S/beta thalassemia.1,2,7
Currently, the use of transcranial Doppler (TCD), a noninvasive ultrasound method that measures the velocity and flow alteration of intracerebral vessels, is considered a sensitive tool for the identification of ischemic stroke risk.1
The association between stroke, coexistence of alpha-thalassemia(del α-3,7) (α-thal), and cluster haplotypes of the beta globin βS is variable in the literature. Studies published with data from Rio de Janeiro,4,8 MG,9 and Sao Paulo4,9 show controversial results. Some studies have reported that the coexistence of α-thal would help reduce the risk of stroke,9–12 while others did not demonstrate such an association.6,13
These studies have identified characteristics regarding the frequency of different haplotypes, with a high prevalence of individuals homozygous for the Central African Republic (CAR) and Benin haplotypes, reflecting the origin of the flow of African slaves who were received at the time of colonial Brazil.4,9,14 In Jamaica and the United States, the Benin haplotype is much more frequent than the CAR haplotype. Such genetic differences make generalizations of results between regions inappropriate.
The aim of this study was to investigate genetic factors associated with risk of stroke in children with SCD.
MethodBetween 1998 and 2007, 188,916 children born in the region of Zona da Mata Mineira and Vertentes underwent neonatal screening for SCD through the PETN-MG. During this period, 135 children with SCD were referred to a reference public institution for hemoglobinopathies. Of this total, nine died before the start of the project, six children with SCD types SD and S_ were excluded, and ten cases were lost to follow-up, thus totaling 110 children who comprised the study population.
Confirmatory diagnosis of SCD was carried out by hemoglobin electrophoresis on alkaline pH after 6 and 12 months of age, measurement of hemoglobin A2 by chromatography, radial immunodiffusion for fetal hemoglobin (HbF), and molecular analysis of codon 6 of beta globin. Children with SCA, SC, S/b0, and S/b+ were considered for inclusion in the study.
Clinical and laboratory data were extracted from medical records, in the period between the date of enrollment at the institution until December 31, 2013 (end of cohort follow-up), allowing a follow-up of at least five years of the study subjects. Socioeconomic characteristics were obtained by applying the questionnaire from the Anisio Teixeira National Institute of Educational Studies and Research (Instituto Nacional de Estudos e Pesquisas Educacionais Anísio Teixeira [INEP]), plus questions about the caregiver's level of schooling and family income. The study to identify the haplotypes and screening for α-thal was initiated in November 2010, after approval by the funding agency through the Research Program for SUS (health system in Brazil) – PPSUS/Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fundação de Amparo à Pesquisa do Estado de Minas Gerais [FAPEMIG]).
The outcome variable was the presence of stroke (yes or no). The diagnosis of stroke was attained clinically (ischemic stroke or transient ischemic attack) or through additional tests such as transcranial Doppler (TCD) and magnetic resonance angiography (MRA) of cerebral vessels.
The screening for the presence of stroke through TCD has been offered annually to all children being followed with SCD, between 2 and 16 years of age, as recommended by the Brazilian Protocol since 2007 in Belo Horizonte, and since 2012 at the regional unit of this study. The test is performed in all patients with SCD, although there are no established velocity parameters for SCD type SC and S/beta+.1 The assessment of the rate of cerebral blood flow in the great arteries of the circle of Willis was based on the criteria from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study.1
The TCD results were stratified according to the classification proposed by the STOP study, adding the recommendation to include the mean maximal velocity (MMV) in one of the anterior cerebral arteries ≥170cm/s as also representing a high risk of developing ischemic stroke.15 The main assessed arteries were the middle cerebral arteries and their bifurcations, and the distal internal carotid artery. The confirmation of an abnormal test (high risk) was made through double repetition, with an interval of one to four weeks.
Children confirmed as high-risk for ischemic stroke were referred for primary preventive treatment of the event with a hypertransfusion regimen. At the time of the TCD, there were no children undergoing exchange transfusion therapy or being treated with hydroxyurea.
Genetic factors considered in the analysis were:
- 1.
Type of SCD (SCA or another type: SC, S/b0, and S/b+ thalassemia) with diagnosis confirmed according to the abovementioned methodology;
- 2.
Gender (male or female);
- 3.
Presence or absence of the mutation for the α-thal gene;
- 4.
Identification of the haplotype (CAR or non-CAR).
To determine the deletion mutation for α-thal and haplotypes, 5mL of whole blood were collected in tubes with ethylenediaminetetraacetic acid (EDTA) during routine SCD laboratory monitoring. The extraction and quantitation of genomic DNA was performed using the commercial QIAamp DNA Blood Mini Kit (QIAGEN®, USA) and Invitrogen® (Invitrogen Corporation®, USA), with restriction enzymes (BioLabs Inc®, USA) according to manufacturer's instructions. The analyses were performed at the research laboratory of the institution.
The identification of the haplotypes was performed using polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP) analysis according Sutton's16 protocol. A commercial kit Multiplex PCR (QIAGEN®, USA) was used for PCR reactions and α−3.7 mutation and deletion screening. The identity of each deletion was obtained by determining the size of the amplified fragment in each reaction. As any deletion removes part or the entire α-2 globin gene, its amplification, together with the amplification of a deletion allele, indicates that the mutation is heterozygous. As positive control for successful DNA amplification, this study used a 2350bp segment, related to the nontranscribed region 3 of the LIS-1 gene (platelet factor) located on chromosome 17p13.3. Other mutations were not screened, considering that in Brazil the deletion that leads to α-thal is the type (3.7).9
The primer sequences were verified against the information available at NCBI (National Center for Biotechnology Information) using the BLAST (Basic Local Alignment Search Tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) tool.
In addition to the described genetic factors, the association between fetal Hb and stroke was assessed. The relative concentrations of fetal Hb in lysed red blood cells (RBCs) was determined at 5 years of age and their value was stratified according to Steinberg17 as fetal Hb ≥10% or fetal Hb <10%.
The children and their parents/guardians were previously informed about the importance of this study, and authorized their participation by signing an informed consent.
The statistical analysis of the associations was carried out through the Chi-squared test, considering a significance level of 5%, using the program SPSS Statistics® 14.0 (IBM Corporation, Somers, NY, United States).
The study was approved by the Research Ethics Committee on 10/9/2009 under No. 245 and is consistent with the provisions of Resolution 466/12 of the National Health Council/Ministry of Health and the Code of Medical Ethics of 1988 (Article 122-130).
ResultsOf the 110 children, 52.7% were males. The mean age at end of the follow-up was 11.2 years, with a standard deviation of 2.84.
Of the study population, 60% were patients with SCA, 33.6% had SC genotype, and 6.4% had S/beta thalassemia (two children S/b0 and five with S/b+). The incidence of SCA was one case per 2857 live births and one case of SCD type SC for every 5128 live births. The family income was up to two minimum wages (MW) in 72.8% of cases, and the mother's level of schooling, the primary caregiver, was complete elementary school in 60.9% of cases.
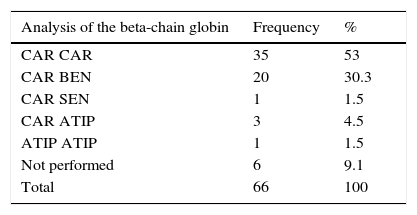
The identification of the haplotypes showed the presence of the CAR gene in the majority of patients (89.2%), reflecting the origin of the African-descent population evaluated in this study. The presence of atypical genes such as Senegal and Cameroon was identified, previously only described in the Northeast of Brazil. It was not possible to define the haplotype in only one child. Seven children (6.4%) did not undergo genetic testing due to refusal or failure to attend blood sample collection within the stipulated time for research. Of the 66 children with SCA, 89.4% had at least one CAR allele (Table 1).
The analysis of the α-globin gene deletion showed an incidence of α-thal of 30.9% of individuals. 26.4% of children were identified with deletion of one gene and 4.5% with deletion of two genes. In this population, no children were identified with deletion of three genes. In relation to SCA, 22.7% of children had deletion of one gene and 7.6% had deletion of two genes for α-thal.
A total of 19 children were identified with stroke, which represented an incidence of 17.2%. Of these, three cases were identified through MRI performed after the clinical manifestation of stroke. Sixteen cases were identified by TCD, 13 of whom showed elevated rates of cerebral blood flow and areas of cerebral ischemia identified at the brain MRA. The increased flow velocity in the right middle cerebral artery and its stenosis were the most prevalent alterations (eight cases). The incidence of stroke among children with SCA was 27.3%. There were no cases of stroke in children SC and S/b+. The mean age at first stroke was 7.7 years, with a minimum age of 6 months and maximum of 15 years.
Table 2 describes the factors associated with stroke. In the assessed sample, only one child with S/beta0 thalassemia had a stroke. Children with SCA had a 12-fold greater risk for stroke than those with other types of SCD (p=0.001). The incidence of stroke among boys was 24.1%, compared to 9.6% among girls (p=0.044). Other factors showed no significant association with stroke.
Frequency of factors associated with stroke in individuals with sickle cell disease.
| Factors | Absolute frequency and percentage of variables | Absolute frequency and percentage of presence of stroke | p-Value | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Type of hemoglobinopathy | 0.001 | ||||
| SCA | 66 | 60 | 18 | 27.3 | |
| S/beta0, S/beta+, SC | 44 | 40 | 1 | 2.3 | |
| Gender | 0.044 | ||||
| Female | 52 | 47.3 | 5 | 9.6 | |
| Male | 58 | 52.7 | 14 | 24.1 | |
| Alpha thalassemiaa | 0.164 | ||||
| Present | 34 | 32.1 | 3 | 8.8 | |
| Absent | 72 | 67.9 | 14 | 19.4 | |
| Haplotypesa | 0.116 | ||||
| CAR | 91 | 89.2 | 17 | 18.7 | |
| Non-CAR | 11 | 10.8 | 0 | 0.00 | |
| Fetal hemoglobin | 0.293 | ||||
| (≥10%) | 46 | 41.8 | 10 | 21.7 | |
| (<10%) | 64 | 58.2 | 9 | 14.1 | |
| Family incomeb | 0.432 | ||||
| Up to 2MW | 80 | 74 | 12 | 15 | |
| >2MW | 28 | 26 | 6 | 21.4 | |
| Maternal level of schoolingb | 0.329 | ||||
| Incomplete elementary school | 41 | 38 | 5 | 12.2 | |
| Complete elementary school | 67 | 62 | 13 | 19.4 | |
| Paternal level of schoolingb | 0.491 | ||||
| Incomplete elementary school | 56 | 51.8 | 8 | 14.3 | |
| Complete elementary school | 52 | 48.2 | 10 | 19.2 | |
SCA, sickle cell anemia; CAR, Central African Republic; MW, minimum wage in Brazil.
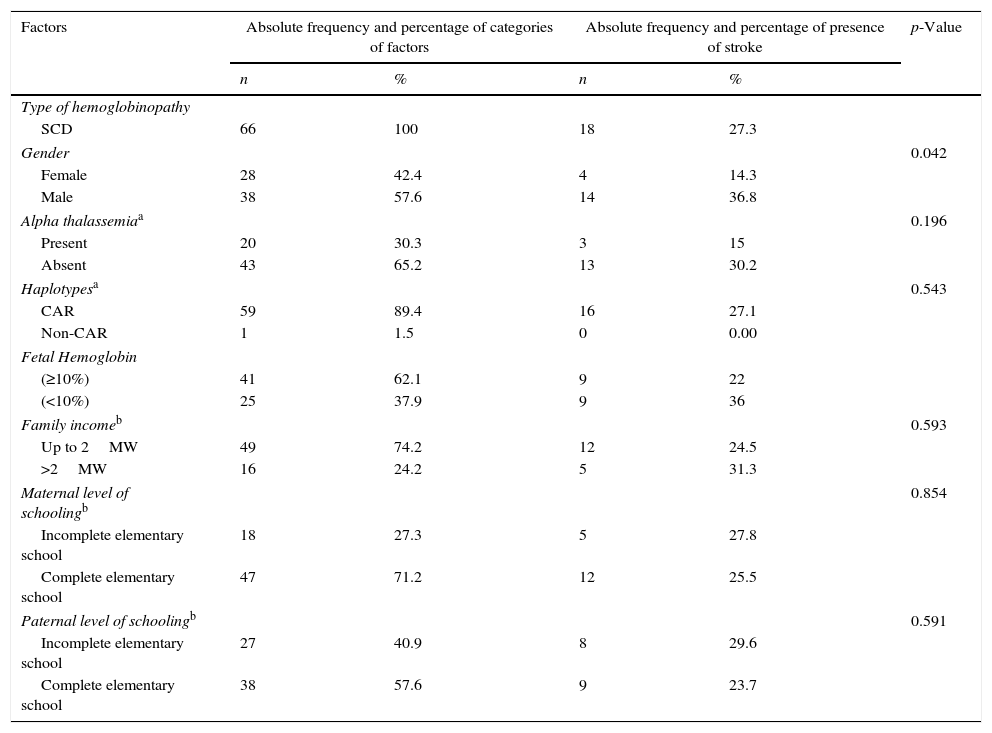
Children with SCA are more susceptible to stroke. For this reason, this group was considered for assessment of the association of stroke with the other factors. The results depicted in Table 3 show that only the variable gender was significantly associated with the outcome (p=0.042).
Frequencies of factors associated with stroke in individuals with sickle cell anemia.
| Factors | Absolute frequency and percentage of categories of factors | Absolute frequency and percentage of presence of stroke | p-Value | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Type of hemoglobinopathy | |||||
| SCD | 66 | 100 | 18 | 27.3 | |
| Gender | 0.042 | ||||
| Female | 28 | 42.4 | 4 | 14.3 | |
| Male | 38 | 57.6 | 14 | 36.8 | |
| Alpha thalassemiaa | 0.196 | ||||
| Present | 20 | 30.3 | 3 | 15 | |
| Absent | 43 | 65.2 | 13 | 30.2 | |
| Haplotypesa | 0.543 | ||||
| CAR | 59 | 89.4 | 16 | 27.1 | |
| Non-CAR | 1 | 1.5 | 0 | 0.00 | |
| Fetal Hemoglobin | |||||
| (≥10%) | 41 | 62.1 | 9 | 22 | |
| (<10%) | 25 | 37.9 | 9 | 36 | |
| Family incomeb | 0.593 | ||||
| Up to 2MW | 49 | 74.2 | 12 | 24.5 | |
| >2MW | 16 | 24.2 | 5 | 31.3 | |
| Maternal level of schoolingb | 0.854 | ||||
| Incomplete elementary school | 18 | 27.3 | 5 | 27.8 | |
| Complete elementary school | 47 | 71.2 | 12 | 25.5 | |
| Paternal level of schoolingb | 0.591 | ||||
| Incomplete elementary school | 27 | 40.9 | 8 | 29.6 | |
| Complete elementary school | 38 | 57.6 | 9 | 23.7 | |
SCD, Sickle cell disease; CAR, Central African Republic; MW, minimum wage in Brazil.
The most severe complication of SCA is stroke, one of the leading causes of death in both children and adults. Despite its high incidence, causes and factors that increase the risk of stroke are not completely known. Several lines of evidence suggest that a genetic signature could influence the development of strokes and that the combined effect of these genes can affect the severity of SCD.2,6,9,15,18,19
Flanangan et al. demonstrated a significant association between α-thal and the genetic polymorphisms (single-nucleotide polymorphisms [SNPS]) ADCY9 rs2238432 in stroke reduction; three SNPs (ANXA2 rs11853426, TEK rs489347, and TGFBR3 rs284875) were significantly associated with an increased risk of stroke; the deficiency of glucose-6-phosphate dehydrogenase and the haplotypes were not related to increased or reduced risk. Belisário et al. described an increase in the risk of stroke in patients that express TNF-alpha (−308G>A), but did not show a significant association with the expression of VCAM-1 polymorphism (c.1238G>C), and associated α-thal with reduced risk of stroke. These results suggest that the association between stroke and polymorphisms remains controversial.1,2,9,15,20–22 The present study assessed the associations between stroke, α-thal, and haplotypes, and no significant results were found, which could be due to the small size of the study population. In this study there were no reports of stroke among children with SCD type SC and S/beta+ thalassemia, data consistent with the Cooperative Study Group in SCD.1
It is worth mentioning that the population of this study consisted of all children diagnosed with SCA (n=66) in the area covered by the health institution that encompasses a population of 730,264 inhabitants [IBGE (Brazilian Institute of Geography and Statistics) Census 2010] Sarnaik and Ballas11 emphasized the importance of multicenter studies with large numbers of patients with and without stroke, to determine the implication of genetic marker studies on the morbidity and mortality in SCD.
The incidence of SCA in this study was one case for every 2857 live births and one case of SCD type SC for every 5128. According to the Ministry of Health, the incidence of SCD detected in the PETN was: Bahia, 1: 650; Rio de Janeiro, 1: 1300; Pernambuco, Maranhão, Minas Gerais, and Goiás: 1: 1400; Espírito Santo, 1: 1800; Sao Paulo, 1: 4000; Rio Grande do Sul, 1: 11,000; Santa Catarina and Parana, 1: 13,500.19
Bezerra et al.5 genetically characterized a cohort of 74 children with SCA in Pernambuco and showed that approximately 65% of patients had the CAR/CAR haplotype, a frequency greater than that found in other states in northeastern Brazil and higher than the 53% found in our study.
This study did not identify the Benin/Benin haplotype among children with SCA, similar results to those found in Rio de Janeiro.13,14 Previous reports showed that haplotypes are associated with an increased risk of stroke.2,11,23,27 According to the present data, the presence of the CAR haplotype (present in 89.2% of cases) was not associated with the development of stroke. These results are similar to those of Flanangan et al.22 and Loghetto,24 who found no significant correlation between the haplotypes and stroke. In 2011, Domingos et al.12 studied a population in Pernambuco different from the one studied by Bezerra et al.,5 with 261 patients, of whom 67 had stroke, and found no association between haplotypes and stroke.
In Brazil, the type of deletion that leads to α-thal is almost exclusively the (−3.7) type.9 The incidence of α-thal found in individuals with SCA in this study was 30.3%, considering the deletion of one and two genes. These data were similar to those found in Salvador (28.2%) and higher than those found by Figueiredo, which was 18.8% in São Paulo.4,9
Several studies12,15,22 have shown the preventive effect of α-thalassemia on the development of stroke in children with SCA. Belisario et al.9 and Hsu et al.10 reported that the frequency of α-thal was significantly higher in patients with SCA with no alterations at the TCD, suggesting molecular protection from the development of stroke. Differently from the present analysis, these markers showed no significant association with stroke risk, results also found by Silva Filho13 and Sommet et al.6 These findings suggest a possible genetic heterogeneity among populations, which may explain the differences among the results of the studies. The manual of SCD conduct and management, “Evidence-Based Management of Sickle Cell Disease” of the National Institutes of Health (NIH), published in 2014, does not advise genetic screening for the prevention of stroke.25
Socioeconomic factors, such as family income and the caregiver's level of schooling, are described as social vulnerability risks and determinants of major health problems of the population with SCD.3,26–29 Such associations were not observed in this study. In Minas Gerais, the implementation of a comprehensive care policy for patients with SCD provides continuous and multidisciplinary care, which may have had a positive impact by reducing the socioeconomic effects on the management of the disease (Center for Education and Support for Hemoglobinopathies – MG – www.cehmob.org.br).
The incidence of stroke was significantly higher in children with SCA and males, data already identified in the International Pediatric Stroke Group.30 The analysis of the coexistence of α-thal and haplotypes in this study showed no correlation in the genesis or prevention of stroke. The heterogenicity between previously assessed populations, non-reproducibility between studies, and the need to identify factors associated with stroke in patients with SCA show the importance of new studies, considering this pathology is the most prevalent monogenic disease in the world and a matter of public health concern.
FundingAll laboratory material used in the development of the study, such as DNA extraction kits, PCR reagents, restriction enzymes, tubes, and pipette tips were purchased with the budget provided by FAPEMIG through PPSUS/FAPEMIG (CDS-APQ-01431-10) and the research laboratory sector of Fundação Hemominas. The research fellowships for the undergraduate students involved in the study were provided by FAPEMIG.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Rodrigues DO, Ribeiro LC, Sudário LC, Teixeira MT, Martins ML, Pittella AM, et al. Genetic determinants and stroke in children with sickle cell disease. J Pediatr (Rio J). 2016;92:602–8.
Study carried out at Fundação Hemominas and Universidade Federal de Juiz de Fora (UFJF), Juiz de Fora, MG, Brazil.




