






Inborn Errors of Immunity (IEI), also known as primary immunodeficiencies, correspond to a heterogeneous group of congenital diseases that primarily affect immune response components. The main clinical manifestations comprise increased susceptibility to infections, autoimmunity, inflammation, allergies and malignancies. The aim of this article is to review the literature on combined immunodeficiencies (CIDs) focusing on the diagnosis and treatment and the particularities of the clinical management of these patients.
Source of dataCritical integrative review, aimed to present articles related to primary immunodeficiencies combined with a searchin the PubMed and SciELO databases, with evaluation of publications from the last twenty years that were essential for the construction of knowledge on this group of diseases.
Summary of dataWe highlight the main characteristics of CIDs, dividing them according to their pathophysiological mechanisms, such as defects in the development of T cells, TCR signaling, co-stimulatory pathways, cytokine signaling, adhesion, migration and organization of the cytoskeleton, apoptosis pathways, DNA replication and repair and metabolic pathways. In CIDs, clinical manifestations vary widely, from sinopulmonary bacterial infections and diarrhea to opportunistic infections, caused by mycobacteria and fungi. Neonatal screening makes it possible to suspect these diseases before clinical manifestations appear.
ConclusionsThe CIDs or IEI constitute a complex group of genetic diseases with T-cell involvement. Neonatal screening for these diseases has improved the prognosis of these patients, especially in severe ones, known as SCIDs.
Inborn Errors of Immunity (IEI), also known as primary immunodeficiencies, correspond to a heterogeneous group of congenital diseases that primarily affect the immune response components. The main clinical manifestations are increased susceptibility to infections, autoimmunity, inflammation, allergies and malignancies.1,2
In the latest report by IUIS (International Union of Immunological Societies), published in January 2020, more than 400 different IEIs have been described, with 430 genetic defects identified and classified into 10 groups1,2:
- -
Immunodeficiencies that affect cell and humoral immunity
- -
Combined immunodeficiencies with associated characteristics or syndromes
- -
Predominantly antibody deficiencies
- -
Immune dysregulation diseases
- -
Phagocyte quantitative or functional defects
- -
Innate immunity defects
- -
Autoinflammatory disorders
- -
Complement system deficiencies
- -
Bone marrow failure or insufficiency
- -
IEI phenocopies
Combined immunodeficiencies are characterized by immunological defects that compromise the development or function of T cells.3 Several genetic disorders can lead to this condition and the Severe Combined Immunodeficiencies (SCIDs) are differentiated from the rest of the group because they are caused by complete gene variants, with full penetrance and with catastrophic functional consequences, leading to a greater susceptibility to potentially fatal infections.
Hypomorphic genetic variants lead to a set of immunodeficiencies combined with milder conditions, the “leaky SCID”.2 Another group of patients has syndromic characteristics associated with combined immunodeficiencies, as a consequence of impaired gene function in non-immune cells, such as DiGeorge, CHARGE, and Bloomsyndrome, among others.4
ObjectiveThe objective of this article is to review the literature on combined immunodeficiencies focusing on the diagnosis and treatment and the particularities of the clinical management of these patients in developing countries.
EpidemiologyIn the last decade, a significant increase in knowledge about IEI has been observed, including clinical and molecular aspects and definition of phenotypes. Traditionally considered as “rare diseases” due to an estimated prevalence of 1:10,000 to 1:50,000, with advances in science and discoveries of new diseases, the actual prevalence is more likely to be 1:10,000 to 1:5,000.1
Even with the evident improvement in technology for the diagnosis of IEI, there is still a significant delay in the identification of these diseases in the developing world, specifically in Latin America.5 The Latin American Immunodeficiency Society (LASID) registry shows a total of 1,286 cases of combined immunodeficiencies including SCIDs, leaky SCIDs and those associated with syndromes. This corresponds to 15% of the total IEI in the registry (Table 1).6
Registry of combined immunodeficiencies of the Latin American Society for Immunodeficiencies (LASID).
| Combined Immunodeficiencies | n |
|---|---|
| Severe Combined Immunodeficiencies (SCIDs) | |
| JAK3 deficiency | 1 |
| γcdeficiency (Common γ chain SCID, CD132 deficiency) | 36 |
| IL7R deficiency | |
| CD3D deficiency | 2 |
| RAG1 deficiency | 9 |
| RAG2 deficiency | 2 |
| DCLRE1C deficiency (Artemis) | 4 |
| DNA ligase IV deficiency | 3 |
| Adenosine Deaminase (ADA) deficiency | 19 |
| T-B- SCID with unknown genetic defect | 89 |
| T-B+SCID with unknown genetic defect | 76 |
| Omenn syndrome | 18 |
| TOTAL | 264 |
| Combined immunodeficiencies with a milder picture than SCIDs(Leaky SCID) | |
| MHC class II group A deficiency | 66 |
| ICOS deficiency | 1 |
| CD8 deficiency | 7 |
| ZAP-70 deficiency/ZAP-70 with hypomorphic and activation mutations | 5 |
| Class I MHC deficiency – TAP2 | 1 |
| Class II MHC deficiency | 2 |
| DOCK8 deficiency | 2 |
| IKBKB deficiency | 2 |
| NIK1 deficiency | 1 |
| Moesin deficiency | 1 |
| TOTAL | 88 |
| Combined immunodeficiencies with associated characteristics or syndromic CIDs | |
| Wiskott-Aldrichsyndrome | 172 |
| Ataxia-telangiectasia | 370 |
| Nijmegen breakage syndrome | 2 |
| Bloom syndrome | 10 |
| DiGeorge/velocardiofacial syndrome | 358 |
| CHARGE syndrome | 1 |
| Cartilage-hair hypoplasia | 11 |
| Schimke immuno-osseous dysplasia | 1 |
| Netherton syndrome | 3 |
| Transcobalamin deficiency 2 | 1 |
| Anhidrotic ectodermal dysplasia with immune deficiency (NEMO/ IKBKGdeficiency) | 1 |
| Anhidrotic ectodermal dysplasia with immune deficiency (gain-of-function IKBA mutation) | 3 |
| STAT5b deficiency | 1 |
| TOTAL | 934 |
SCIDs are characterized by profound defects in T- and B-lymphocyte function and a significantly low T-cell count. It affects approximately 1:55,000 newborns and less than a third have a family history. It is not clinically apparent at birth and infectious complications usually appear during the first year of life, being potentially fatal until the age of two, if immune reconstitution is not performed.7,8
It is considered a pediatric emergency and an early diagnosis is essential for successful treatment. With the advent of neonatal screening for SCIDs, there has been an improvement in the prognosis of these patients, since the performance of hematopoietic stem cell transplantation (HSCT) in children without infection results in a 2-year survival in approximately 95% of cases.9
However, in Latin America and in many parts of the world where neonatal screening for SCID is not yet routinely available, the diagnosis is made most of the time with infections and severe complications and referral for definitive treatment at a specialized center is delayed.8
In Brazil, a country of continental dimensions, a pioneering initiative sponsored by the Jeffrey Modell Foundation investigated 60 cases of suspected SCIDs, from 2016 to 2018, from all the regions of the country and diagnosed 25 patients with a median age of 5.5 months at the diagnosis.10
Classification of combined immunodeficiencies or combined IEIWe can classify the Combined IEI according to the altered molecular mechanism:
Defects in the development of T cellsDefects in the development of T cells can result from changes in thymic development or recombination of V (D) J. Changes in thymic development lead to thymic hypoplasia and the T-cell defect can vary from mild to a SCID phenotype in the most severe cases (thymus aplasia).11 Related genes: TBX1 haploinsufficiency caused by the deletion of chromosome 22q11.1 causing DiGeorge syndrome; Autosomal dominant CHARGE syndrome with a mutation in the SEMA3E and CHD7 genes; FOXN1 leading to autosomal recessive SCID.12
The genetic recombination of V (D) J produces genes that encode the T-cell (TCR) and B-cell (BCR) antigen receptor chains. This genetic recombination is important to generate different specificities of receptors that precede contact with different antigens and then a varied repertoire of lymphocyte clones. Alterations in these genes generate severe B- and T-lymphocyte defects, leading to a SCID T-/B-/Natural Killer (NK)+ phenotype. The main disease related to this defect is Omenn Syndrome (characterized by rash, eosinophilia, oligoclonal and autoreactive T cells, variable CD3, which can be >1500). The main genes: RAG 1 e 2; Artemis and DNA-PKcs; DNA ligase IV, the last ones with radio sensitivity and the latter also with syndromic characteristics.13
Defects in TCR signalingSome genetic variants can lead to proximal signaling alterationsmediated by TCR. These diseases lead to a general alteration in the T-cell compartment. They have a decrease in regulatory T cells (TRegs) and an alteration in thymic selection, with the formation of self-reactive T cells, leading to changes in the activation, effector function and formation of T-cell memory. Types of autosomal recessive SCID, with susceptibility to bacteria and viruses plus the autoimmunity are associated, and variants that affect the α β ¿ δ chains of the TCR, with variants in the δ chain being more harmful than in the ¿ chain.14
Other diseases related to TCR signaling defects: CD8α deficiency; CD45 deficiency (SCID with T-/B+/NK+phenotype); ZAP-70 (selective CD8 deficiency with normal CD4 number, but with altered function)15; Calcium influx diseases: ORAI-1 and STIM 1; deficiency of EVER 1 and 2 (verruciform epidermodysplasia caused by HPV infection); bi-allelic variants with loss of function (LOF) in PRKCD (recurrent infections, lupus-like, chronic lymphadenopathy and splenomegaly due to B-cell hyperactivation and proliferation); phosphoinositide signaling diseases (proteins important for regulating T-cell activation and migration): mutations with gain of function (GOF) in the monoallelic germline of PIK3CD/PIK3R1/PTEN leading to immunodeficiency, immune dysregulation and susceptibility to cancer.16
Defects in co-stimulatory pathwaysCo-stimulatory molecules are necessary for the complete activation of the T lymphocyte, effector functions, memory formation and anergy prevention, in addition to stimulation by TCR. These molecules are cell surface receptors that recognize ligands in antigen-presenting cells (APCs) or target cells after infection.
Diseases related to co-stimulatory pathways are: CARMIL2/RLTPRdeficiency (via CD28, induces NF-kB-mediated CADR-11 activation) leading to a decrease in TReg, with an increase in the population of naïve CD4 and CD8 and a decrease in memory populations, lung and skin infections, allergy and susceptibility to EBV; CTLA-4 insufficiency (expressed in TReg and activated T cell, CTLA4 counter regulates CD28-dependent signaling and its deficiency leads to lymphocytic infiltrates, autoimmunity and immunodeficiency, with great clinical variability); ICOS deficiency (selective defect in the formation, migration and function of follicular T cells, T lymphocytes do not produce IL10 and IL17 after stimulation, leading to a defect in the production of antibodies and germinal centers, with susceptibility to infections, hepatosplenomegaly and malignancies); CD40L mutation (X-linked Hyper-IgM syndrome); CD27/CD70 mutation (EBV-associated lymphoproliferative diseases).17
Cytokine signaling defectsThe cytokine signaling pathway ranges from functions of the cytokines themselves and cytokine receptors to JAK and STAT signal mediators (signal transducer and transcription activator). Defects in this pathway lead to immunodeficiency. Some diseases related to cytokine signaling: mutation of the common gamma chain (X-linked SCID with T-/B-/NK- phenotype); mutation in IL7Rα (autosomal recessive T/B+/NK+); mutation in IL2RA (CD25) (IL2Ra combined with IL2Rb and common gamma chain form a high affinity IL2 receptor, causing autosomal recessive immunodeficiency associated with autoimmunity – this defect combines IPEX-like with specific T-cell disease); mutation in IL21/IL21R (recurrent respiratory infection, hepatitis, liver fibrosis, diarrhea, septicemia, B-lymphocyte with decreased class switch and impaired T-lymphocyte response to candida); mutation in JAK3 causing autosomal recessive SCID (JAK1 makes a dyad with JAK3 to trigger signaling via the common gamma chain); STAT 3 dominant negative mutations (Autosomal dominant hyper IgE syndrome); STAT3 GOF (interstitial pneumonia, autoimmune enteropathy, arthritis, lymphadenopathy, T-cell leukemia).18
Defects in cytoskeleton adhesion, migration and organizationThe cells of the immune system require extreme mobility and interaction with several cell types so they can migrate to the sites of infection, receive activation signals and exercise an effector function. Some of the diseases related to these functions are: DOCK2 (early onset of bacterial and viral infections, normal NK with poor function); DOCK8 (LOF; sinopulmonary infections; bacterial and viral skin infections; high IgE with severe atopy and anaphylaxis); WASP(protein encoded by the WAS gene –depending on the mutation it causes: Wiskott-Aldrich syndrome, congenital neutropenia and X-linked thrombocytopenia); CXCR4 (WHIM syndrome: neutropenia, warts caused by HPV, hypogammaglobulinemia, recurrent infections); Leukocyte adhesion deficiency (LAD) I and III.
Defects in apoptosis pathwaysAfter infection resolution, the clonal expansion of B and T lymphocytes should be reversed to homeostasis. The main related IEIs have alterations in the apoptosis-inducing signaling complexes: FAZ/FASL/Caspase8/caspase10/FADD leading to APLS (non-malignant lymphadenopathy, double negative cells [CD4- / CD8- / αβ +] and autoimmune cytopenias).19 Another disease that has a FAZ-related pathogenesis is the X-linked proliferative syndrome, with a LOF mutation in XIAP (recurrent infections, Epstein-Barr virus viremia, inflammatory bowel disease and hemophagocytic lymphohistiocytosis).
Defects in DNA replication and repairB and T lymphocytes undergo periods of rapid DNA proliferation and replication with a high chance of damage, and there pair of this DNA damage is essential for the cell cycle. ATM mutations (LOF) lead to ataxia-telangiectasia (ataxia, telangiectasia, immunological defects, malignancy).20
Defect in metabolic pathwaysDuring the ontogeny and life cycle of the T lymphocyte, the metabolic properties of the T lymphocyte are extremely important. Defects in this pathway cause: Reticular dysgenesis (AK2 mutation leading to SCID with early defect in the myeloid lineage cells –absence of granulocytes, severe lymphopenia, thymicand lymph nodehypoplasia); ADA deficiency (cause of SCID, but clinical variety depends on the amount of residual ADA activity)21; PNP (autosomal recessive SCID, bacterial and viral infections, absence or small type, autoimmune disease).
In the case of SCIDs, we can classify the phenotypes according to the presence or absence of B, T lymphocytes and NK cells, as shown in Fig. 1.
Clinical manifestations
The main clinical manifestation of IEI is the susceptibility to infections. The patient with recurrent infections, unusual infections or infections that are difficult to treat should be investigated.22 In combined immunodeficiencies, clinical manifestations show great variability, from sinopulmonary bacterial infections and diarrhea to opportunistic infections, by mycobacteria and fungi and vaccinal reaction with loco-regional to systemic symptoms (by BCG – Bacillus Calmette-Guérin), being more severe in SCIDs, usually fatal in the early years of life, if left untreated.23
Compared to other IEIs, infections start before 6 months of age and are normally associated with low weight-height gain in SCIDs. Autoimmune manifestations, such as cytopenias, can occur and genotypes that show congenital abnormalities at birth are rare.
Biochemical diagnosisWhen the diagnosis of combined immunodeficiency is suspected after clinical evaluation, initial laboratory tests should be requested (Table 2 and Fig. 2). The patient's complete blood count (CBC) gives clues of immunological alterations. The assessment of the absolute neutrophil and lymphocyte count should be performed according to the patient's age.24 HIV diagnosis should be ruled out in all patients.
Scheme for the evaluation and management of patients with combined IEI.
| Clinical history including infections, family history and consanguinity |
|---|
| Detailed physical examination: visceromegaly, rash or erythroderma, congenital abnormalities such as microcephaly |
| Serum tests: CBC, immunoglobulin levels, t, B and NK lymphocyte immunophenotyping, TREC/KREC |
| Evaluation of memory cells in immunophenotyping; Lymphoproliferation with PHA |
| Assessment of autoimmune cytopenias, liver function |
| PCR for CMV, Herpes virus, EBV and HIV; maternal serology can be performed |
| If the mother is breastfeeding: PCR for CMV for the mother – if negative, encourage breastfeeding. IF positive, suspend |
| Vaccinal evaluation to date and contraindicate live vaccines |
| Contact persons: do not vaccinate with oral polio; avoid contact if sick |
| Start prophylaxis: Sulfa, fluconazole and acyclovir. If person received BCG, start isoniazid |
| Start human immunoglobulin replacement therapy, maintain IgG>800mg/dL |
| Nutritional support |
| Administer Palivizumab during RSV season |
| HLA collection for BMT assessment |
| If there are characteristics of DiGeorge or heart disease, perform CGH-array for chromosome 22q11 |
| Evaluate NGS/genetic panel/exome according to SCID phenotype |
| If there is non-SCID lymphopenia, assess differential diagnoses: alpha-fetoprotein measurement (>7 months of life) |
NGS, next generation sequencing; PHA, phytohemagglutinin; SCID, severe combined immunodeficiency.

The evaluation of specific immunological parameters should be performed through the measurement of immunoglobulins (IgA/IgG/IgM/IgE), vaccinal response after 6 months of life (when maternal antibodies transferred via the placenta decrease) and measurement of the larger leukocyte subtypes by flow cytometry (immunophenotyping of CD3/CD4/CD8/CD19 and NK).More specific flow cytometry evaluations for assessing naïve and memory cells are important and should also be assessed according to the patient's age,24 with the lack of naïve lymphocytes being a major predictor of SCID. Lymphocyte function can be measured by lymphoproliferation after stimulation with phytohemagglutinin (PHA).22
Molecular diagnosisThe last diagnostic modality is the identification of a genetic variant of which product is involved in immunity. After the adoption and evolution of genomic technologies, such as next generation sequencing (NGS), the number of diseases and genetic defects related to IEI increased considerably. Genetic sequencing in SCIDs is very important, both for the definitive diagnosis and also for pre-HSCT conditioning and family genetic counseling.25
Pathogenic DNA variants, excluding copy number variation (duplications or deletions), can be identified by sequencing a single gene or by sequencing the exome.26 Variants that lead to the complete deletion of the encoded protein are associated with increased immunodeficiency severity, and hypomorphic mutations that preserve some function of the protein result in leaky SCIDs.27
There are monogenic diseases caused by variants present in non-coding regions (introns) that have regulatory elements, such as promoters or non-coding genes such as the microRNA that regulates other IEI-related proteins, which are not evaluated by the exome. Evaluation of the genome associated with transcriptome, proteome and epigenome is possible and may be necessary – the use of the exome combined with RNA sequencing has provided some answers.25
Neonatal screeningThe population-based neonatal screening allows the early identification of asymptomatic babies with a variety of severe
diseases, for which effective treatment exists and where early diagnosis and intervention prevent severe sequelae.28
Until recently, it was not possible to identify babies with IEI before the onset of clinical symptoms and with complications of severe and prolonged infection. Advances in molecular biology and biotechnology have allowed the identification of babies with severe forms of IEI manifested by T- and/or B-cell lymphopenia.29
Neonatal screening programs for IEI, including T-cell receptor excision circles (TRECs) and kappa recombination excision circles (KRECs) are screening approaches that assess the maturation of the T-cell (TCR) and B-cell (BCR) receptor (Fig. 3). For this sequence of events, these circles are ejected into the bloodstream by primary gene rearrangement or also the so-called VDJ recombination.30

Simplified TREC collection and detection scheme. Adapted from Somech and Etzioni.28
V(D)J recombination occurs in the primary lymphoid organs (bone marrow for B cells and thymus for T cells) and semi-randomly rearranges the gene segments V (variable), J (joining) and, in some cases, D (diversity). The process results in new amino acid sequences in the antigen-binding regions of Immunoglobulins and TCRs that allow recognizing antigens from virtually all pathogens, including bacteria, viruses and parasites.31
Although the identification of babies with SCIDs was the intended goal of TREC-based neonatal screening programs, it became evident that, in addition to this disorder, the assay would also identify babies with T-cell lymphopenia due to other primary and secondary causes. For example, low levels of TREC have been detected in individuals with 22q deletion, association with CHARGE syndrome and Trisomy 21. Moreover, babies with forms of IEI other than SCID may have low TREC, for instance, in ataxia-telangiectasia and in combined immunodeficiencies (CIDs). To date, in prospective pilot studies, many cases of CID without an identifiable molecular cause have been detected using TREC and these patients require clinical characterization and long-term follow-up.30
Some limitations can happen when the molecular defect is downstream of the T-cell receptor rearrangement, so it cannot be detected. This includes Zap70 deficiency, MHC Class II deficiency and some cases of ADA. Defects in T-cell function, despite a quantitatively normal number of T cells, will also not be detected by TREC.28
Prophylaxis for combined immunodeficienciesa) Immunoglobulin replacement therapy
Immunoglobulin replacement therapy is the most important treatment for IEIs with antibody production defects. The predominantly antibody defects, combined defects of T and B cells and syndromes associated with immunodeficiencies stand out, according to the latest classification published in early 2020.2
The usual replacement dose is 400 to 600mg/kg/month for intravenous (IV) presentation or 100–150mg/kg/week for the subcutaneous (SC) presentation. Higher doses, called immunomodulatory (1–2g/kg of weight), are used in autoimmune manifestations.32
The most frequent adverse effects are headache, malaise, nausea, tremors, fever, chest pain and coagulation changes, particularly with the intravenous administration, and are directly related to the dose and the velocity of administration. With the subcutaneous presentation, systemic adverse effects are much less frequent. More commonly, the SC use is associated with local irritation, which decreases with continuing treatment.33
b) Prophylactic antimicrobials
Susceptibility to infections is an important characteristic ofcombined immunodeficiencies and determines the clinical evolution of patients and, for this reason, antimicrobial prophylaxis are frequently used to prevent infections and their complications.34,35 The choice of prophylactics should take into consideration the type of infectious agent to which the patient is susceptible based on laboratory alterations, their clinical history, the infectious agents isolated from previous infections or that colonize this patient, as well as information obtained from the literature.
Patients with combined immunodeficiencies, especially severe ones (SCIDS) are susceptible to all types of infections and infectious agents. Given the severity and high mortality in the first years of life, a portion of these patients should receive definitive treatments with hematopoietic stem cell transplantation and thymus transplantation in patients with complete DiGeorge syndrome.
However, while awaiting definitive treatment, these patients should receive antimicrobial prophylaxis with regular immunoglobulin replacement, palivizumab during the respiratory syncytial virus season and prophylactic antibiotics for P. jirovecii, herpes family viruses and Candida.35 The antimicrobials used and their doses are shown in Table 3. Patients who received BCG vaccine and do not have adverse reactions should also receive prophylaxis with isoniazid and anti-tuberculosis treatment in case of BCG complications (Tables 2 and 3).36
Prophylactic antimicrobials suggested for patients with severe combined immunodeficiency awaiting definitive therapy.
| Prophylaxis | Medication | Start | Comments |
|---|---|---|---|
| Pneumocystosis | TMF-SMX (5mg/kg/day) | 1 month | Liver function |
| Herpes andvaricella zoster | Acyclovir 20mg/kg/dose3×/day | At diagnosis | Kidney function |
| RSV | Palivizumab | 1 month | Seasonality |
| General infections | Human immunoglobulin | 1 month | Serum IgG level |
| Fungalinfections | Fluconazole 6mg/kg/day | 1 month | Liver function |
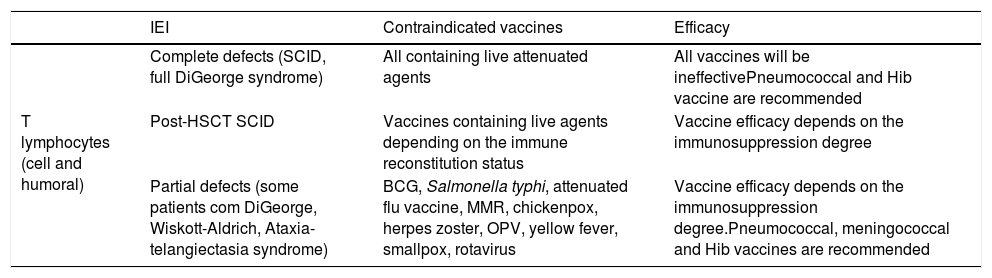
Vaccination is an efficient tool in preventing infections and represents a major advance in public health. However, patients with IEI, depending on the type and severity of the immunological compromising, may experience an absent or reduced response to vaccination.8 Additionally, some patients with IEI may be susceptible to the occurrence of infectious complications when submitted to vaccines containing live attenuated agents, as already mentioned in BCG vaccination (Table 4).37 Therefore, decisions about indicating or contraindicating vaccines to a patient with IEI must take into account the immunological condition determined by the underlying disease, which may be aggravated by complications or the use of medications. As a general recommendation for decisions related to the vaccination of immunocompromised patients, we should always carefully evaluate risks and benefits, consider that inactivated vaccines are generally safe, and that those that contain live attenuated agents are, in general, contraindicated.38
Recommendations for the vaccination of patients with combined immunodeficiencies.a
| IEI | Contraindicated vaccines | Efficacy | |
|---|---|---|---|
| T lymphocytes (cell and humoral) | Complete defects (SCID, full DiGeorge syndrome) | All containing live attenuated agents | All vaccines will be ineffectivePneumococcal and Hib vaccine are recommended |
| Post-HSCT SCID | Vaccines containing live agents depending on the immune reconstitution status | Vaccine efficacy depends on the immunosuppression degree | |
| Partial defects (some patients com DiGeorge, Wiskott-Aldrich, Ataxia-telangiectasia syndrome) | BCG, Salmonella typhi, attenuated flu vaccine, MMR, chickenpox, herpes zoster, OPV, yellow fever, smallpox, rotavirus | Vaccine efficacy depends on the immunosuppression degree.Pneumococcal, meningococcal and Hib vaccines are recommended |
BCG, Bacillus Calmette-Guérin; Hib, Haemophilus influenzae; SCID, severe combined immunodeficiency; HSCT, hematopoietic stem cell transplantation; MMR, measles-mumps-rubella; CGD, chronic granulomatous disease.
HSCT is currently the curative treatment of choice for patients with severe and lethal forms of IEI.
The aim of HSCT is to replace the patient’s deficient immune system by an effective immune system from a healthy donor. The success of HSCT is directly associated with the availability of a donor (degree of compatibility between donor and recipient in the HLA– human leukocyte antigen– system), the characteristics and particularities of each disease to be treated, and the patient’s clinical condition at the time of transplantation.
The donor for the transplant is sought for compatibility in the HLA system and can be found within the family (related) or in the voluntary donor bank (unrelated). The cell sources used include bone marrow, peripheral blood or umbilical cord stem cells. The conditioning regimen is used for the patient's immunosuppression, preventing transplant rejection, with chemotherapy and/or radiation therapy usually being used.39 There are different types of conditioning regimens, chosen according to the patients' clinical and immunological characteristics. Some new studies also consider genetic variants to decide about the conditioning. Briefly, it can be divided into RIC (Reduced Intensity conditioning), which is a lighter conditioning, and MAC (Myeloablative conditioning), this one being more severe.40
The donor stem cells are infused through a central venous catheter. The recovery of the leukocytes from the donor cells (>500cells/mm3) is called graft “taking” and occurs approximately 2–4 weeks after the infusion. The main complications associated with transplantation include: toxicity directly associated with the chemotherapy used (veno-occlusive liver disease, mucositis, hemorrhagic cystitis); infections (bacterial, viral or fungal); and graft-versus-host disease (caused by the reactivity of donor cytotoxic T cells against the recipient’s cells – mainly affecting the skin, gastrointestinal tract and liver). Long-term complications include chronic graft-versus-host disease, endocrinological problems and infertility.39
Gene therapyGene therapy can be understood as the capacity for genetic improvement through the correction of altered genes or site-specific modifications, which target therapeutic treatment.
Didactically, three techniques are available for the cure of IEI, which are: gene addition/insertion, gene editing, and gene silencing (most used in IEI with gain of function). The first is most often used and consists of recombinant DNA technology, in which the gene of interest or healthy gene is inserted into a vector, which can be plasmodial, nano-structured or viral, with the latter being the most often used, due to its efficiency in invading cells and introducing their genetic material into them.41
Although several protocols have been successful, the gene therapy process remains complex and many techniques need improvement. There is also the important question of the cell type targeted by the gene therapy, which is currently divided into two major groups: germline gene therapy (sperm and egg) and somatic cell gene therapy (therapeutic genes are transferred into somatic cells of a patient).41
In the 1980s, a region with an unusual pattern was identified in the genome of the Escherichia coli bacterium, in which a highly variable sequence was intercalated with a repeated sequence with no known function. In 2005, it was postulated that the variable sequences were of extrachromosomal origin, acting as an immunological memory against phages and plasmids, initiating the then unknown Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and Cas (Associated Proteins) system, which has been shining since 2012 as one of the main biotechnological tools for genome editing.41
Coming from the immune-adaptive system of prokaryotes, this mechanism recognizes the invading genetic material, cleaves it into small fragments and integrates it into its own DNA. When a second infection by the same agent occurs, the following ensues: transcription of the CRISPR locus, mRNA processing and creation of small fragments of RNA (crRNAs), which form complexes with Cas proteins, and these recognize foreign nucleic acids and finally destroy it. Based on this natural mechanism, the CRISPR technique was developed, which makes it possible to edit target-specific DNA sequences in the genome of any organism through the exclusive action of only 3 molecules: the nuclease (Cas9), responsible for the cleavage of double-stranded DNA; a guide RNA, which guides the complex to the target; and the target DNA.41,42
Gene therapy is now being tested as a therapeutic option for an increasing number of diseases, based primarily on the successful treatment of patients with IEI in the past two decades, including severe combined immunodeficiency (SCIDs) and Wiskott-Aldrich syndrome. The field has developed from the use of gamma-retroviral vectors to more sophisticated lentiviral platforms that offer an improved biosafety profile, in addition to greater efficiency in the transfer of genes from hematopoietic stem cells.42
ConclusionsCombined immunodeficiencies or combined IEI constitute a complex group of genetic diseases with T-cell involvement. Neonatal screening for these diseases has improved the prognosis of these patients, especially in SCIDs. Developing countries still do not routinely have these tests for the entire population, resulting in delayed diagnosis.
The care related to the replacement of human immunoglobulin and the use of prophylactic antimicrobials contribute to the reduction of infections and their complications. Major advances related to hematopoietic stem cell transplantation and gene therapy will allow the definitive treatment of many patients with IEI, especially for combined immunodeficiencies.
Conflicts of interestThe authors declare no conflicts of interest.




