


this study aimed to investigate the cognitive and behavioral profiles, as well as the psychiatric symptoms and disorders in children with three different genetic syndromes with similar sociocultural and socioeconomic backgrounds.
Methodsthirty-four children aged 6 to 16 years, with Williams-Beuren syndrome (n=10), Prader-Willi syndrome (n=11), and Fragile X syndrome (n=13) from the outpatient clinics of Child Psychiatry and Medical Genetics Department were cognitively assessed through the Wechsler Intelligence Scale for Children (WISC-III). Afterwards, a full-scale intelligence quotient (IQ), verbal IQ, performance IQ, standard subtest scores, as well as frequency of psychiatric symptoms and disorders were compared among the three syndromes.
Resultssignificant differences were found among the syndromes concerning verbal IQ and verbal and performance subtests. Post-hoc analysis demonstrated that vocabulary and comprehension subtest scores were significantly higher in Williams-Beuren syndrome in comparison with Prader-Willi and Fragile X syndromes, and block design and object assembly scores were significantly higher in Prader-Willi syndrome compared with Williams-Beuren and Fragile X syndromes. Additionally, there were significant differences between the syndromes concerning behavioral features and psychiatric symptoms. The Prader-Willi syndrome group presented a higher frequency of hyperphagia and self-injurious behaviors. The Fragile X syndrome group showed a higher frequency of social interaction deficits; such difference nearly reached statistical significance.
Conclusionthe three genetic syndromes exhibited distinctive cognitive, behavioral, and psychiatric patterns.
investigar o perfil cognitivo e comportamental, sintomas e transtornos psiquiátricos em crianças com três diferentes síndromes genéticas, com antecedentes socioculturais e socioeconômicos semelhantes.
Métodostrinta e quatro crianças, entre 6 e 16 anos, com as síndromes de Williams-Beuren (n=10), de Prader-Willi (n=11) e do X-Frágil (n=13), dos ambulatórios de Psiquiatria Infantil e Genética Médica, foram avaliadas cognitivamente pela Escala Wechsler de Inteligência para Crianças (WISC-III). Posteriormente, o QI total, o QI Verbal, o QI de Execução, os escores ponderados dos subtestes e a frequência de sintomas e transtornos psiquiátricos foram comparados entre as síndromes.
Resultadosdiferenças significativas foram encontradas entre as síndromes quanto ao QI Verbal e os subtestes verbais e de execução. A análise Post-hoc demonstrou que os escores dos subtestes vocabulário e compreensão foram significativamente superiores na síndrome de Williams-Beuren em relação às síndromes de Prader-Willi e do X-Frágil, e os escores dos subtestes cubos e armar objetos foram significativamente superiores na síndrome de Prader-Willi em relação às síndromes de Williams-Beuren e do X-Frágil. Além disso, houve diferença significativa entre as síndromes quanto às características comportamentais e os sintomas psiquiátricos. O grupo com síndrome de Prader-Willi apresentou maior frequência de hiperfagia e comportamentos autolesivos. Já o grupo com síndrome do X-Frágil apresentou maior frequência do déficit da interação social. Esta diferença quase alcançou a significância estatística.
Conclusãoas três síndromes genéticas apresentaram um padrão cognitivo, comportamental e psiquiátrico diferenciado quando foram comparadas entre si.
Intellectual disability (ID), the current term for mental retardation, is one of the most commonly observed neuropsychiatric disorders that impairs social functioning and adaptive behavior of children and adolescents.1 In underdeveloped countries, the prevalence of ID is almost two times higher than in developed countries.2
Common causes of ID are genetic diseases, problems during pregnancy or birth, birth defects that affect the brain, and problems during infancy, childhood, and adolescence, such as injuries, diseases, or brain abnormalities.3 In underdeveloped and developing countries, malnutrition, socio-cultural deprivation, and poor healthcare are also factors frequently associated with ID.4
Patients with ID present higher risk for psychiatric disorders than the general population. The rate of psychiatric disorders in this population ranges from 30% to 50%.4
Despite the high prevalence of ID and strong association with psychiatric disorders, mental health professionals often fail to give proper attention to ID.5,6 When caring for less prevalent conditions in mental healthcare, such as genetic syndromes with ID,7 clinicians frequently ignore their specific cognitive, behavioral, and psychopathological characteristics.
Three genetic syndromes featuring ID have been receiving increasing attention by specialists in the care of children with genetic syndromes due to their diverse expression of cognitive and behavior characteristics: Williams-Beuren syndrome (WBS), Prader-Willi syndrome (PWS), and Fragile X syndrome (FXS).8–10
WBS, a rare neurodevelopmental disorder caused by a submicroscopic deletion on chromosome 7q11.23, is characterized by dysmorphic facial features, elastin arteriopathy, short stature, connective tissue abnormalities, infantile hypercalcemia, and ID.11 Children with WBS usually display high sociability, excessive empathy (which may be inappropriate), anxiety, preoccupations and fears, impulsivity, inattention, sadness and depression, generalized anxiety disorder, phobias, and attention deficit hyperactivity disorder.7,12 Relatively good language skills and verbal short-term memory, and a marked deficit in visuospatial skills have been described in WBS.8,13
PWS, a genetic disorder that results from abnormality or loss of a critical region of chromosome 15q11–13, is characterized by neonatal hypotonia, hyperphagia with eventual obesity, and ID.7 Children with PWS usually have good performance in visuospatial construction tasks,5,9 but present important deficits in mathematics14 and expressive language.15
Individuals with PWS exhibit a distinctive behavioral phenotype, with temper tantrums, stubbornness, and excessive interest in food; as well as obsessive, compulsive, manipulative, oppositional, and defiant behaviors.16 The psychiatric features commonly reported in PWS are obsessive-compulsive disorder, depression/mood disorder, psychosis, and self-injurious behaviors (skin picking).7
FXS, a disorder caused by an unusually large tri-nucleotide repeat (CGG) expansion in the long arm of the X chromosome, is the most common cause of inherited ID.10 The cognitive profile in FXS includes deficits in executive control and in visuospatial abilities,17 as well as in pragmatic language and morphosyntax, but not in vocabulary.18
Males with FXS present more severe cognitive impairments when compared to females with the same syndrome,19 and frequently manifest behaviors from the autistic spectrum, such as gaze aversion, social avoidance, and stereotypical and repetitive behavior.20
Individuals with FXS often meet criteria for attention deficit hyperactivity disorder, oppositional defiant disorder, enuresis, encopresis, and exhibit isolated symptoms and behaviors that do not always fit into the diagnostic categories employed by Diagnostic and Statistical Manual of Mental Disorders (DSM), such as anxiety and compulsive symptoms, labile mood, irritability, aggressive outbursts, self-injurious behavior, impaired attention, and hyperactivity.21
Although each of these individual genetic syndromes associated with ID have been individually investigated due to their diverse expression of cognitive and behavior characteristics, studies that compare them, enrolling participants from similar social and cultural background and using the same methodology for cognitive and behavior/psychiatric assessments are still scarce.
Thus, the present study aimed to investigate the cognitive profiles and behavioral features, as well as psychiatric symptoms and disorders in children and adolescents with WBS, PWS, and FXS.
MethodsThis was an analytical cross-sectional study that used a convenience sample. All children and adolescents with WBS, PWS, or FXS from the outpatient clinics of the Child and Adolescent Psychiatry and Medical Genetics Department of the University Hospital of the University of Campinas (Unicamp - Campinas, Brazil) were enrolled in this study. Two participants with WBS came from an institution specialized in the care of children with ID (Campinas, Brazil). Considering that WBS is a relatively rare syndrome, this strategy was adopted in order to make the sample size of the three groups comparable.
This study was approved by the Institutional Review Board of the Faculty of Medical Sciences, Unicamp. The sample obtained consisted of 34 children and adolescents aged 6 to 16 years; ten participants had WBS (seven males and three females); 11 participants had PWS (five males and six females); and 13 participants had FXS (12 males and one female). The participants had similar sociocultural and socioeconomic backgrounds (Table 1). Consent forms approved by the Institutional Review Board of Unicamp were signed by the parents.
Sociodemographic characteristics.
| WBS (n=10) | PWS (n=11) | FXS (n=13) | p-value | |
|---|---|---|---|---|
| Age–Mean (SD) | 11.7 (3.6) | 11.1 (2.7) | 12.0 (3.0) | 0.70a |
| Gender | 0.04b | |||
| Male | 7 (70%) | 5 (46%) | 12 (92%) | |
| Female | 3 (30%) | 6 (54%) | 1 (8%) | |
| Type of education | 0.99b | |||
| Special school | 8 (80%) | 9 (82%) | 11 (84%) | |
| Regular school | 2 (20%) | 2 (18%) | 2 (16%) | |
| Family Incomec | 0.98b | |||
| ≤ 2.0 | 4 (40%) | 6 (55%) | 6 (46%) | |
| 2.1-3.0 | 3 (30%) | 3 (27%) | 4 (31%) | |
| > 3.0 | 3 (30%) | 2 (18%) | 3 (23%) | |
| Per capita incomec | 0.74b | |||
| ≤ 1.0 | 8 (80%) | 10 (91%) | 10 (77%) | |
| > 1.0 | 2 (20%) | 1 (9%) | 3 (23%) | |
FXS, Fragile X syndrome; PWS, Prader-Willi syndrome; WBS, Williams-Beuren syndrome.
Significant values in bold.
Children and adolescents with clinical diagnosis of WBS, PWS, or FXS confirmed by cytogenetic exams that were assessed by a clinical psychiatrist from the outpatient clinics of the Child and Adolescent Psychiatry Department were included in the study. Patients who did not develop language, which would prevent the psychological assessment, were excluded.
WBS diagnosis was confirmed through the fluorescence in-situ hybridization technique. All participants with PWS had their diagnosis confirmed by fluorescence in-situ hybridization technique and/or by methylation analysis of the SNRPN gene. All participants with FXS had their diagnosis confirmed by molecular study of the FRAXA mutation, using the Southern blotting technique.
Clinical psychiatrists (EHRV and PD) diagnosed the participants using the fourth edition of the DSM, Text Revision.22 Psychiatric symptoms and diagnoses, behavioral characteristics (e.g., explosiveness, oppositional behavior, hyperphagia), and sociocultural and socioeconomic features (i.e., family income, per capita income, and educational level of the participants and their parents) were obtained by medical chart review before the cognitive evaluation of each participant. These data were previously acquired during psychiatric assessment/anamnesis with participants’ caregivers, through a structured protocol.
The Brazilian version of Wechsler Intelligence Scale for Children (WISC-III), third edition,23 was applied by a psychologist (LFLP) after the psychiatric assessment of each children and adolescent. The WISC-III is an individually administered measure of intelligence intended for children aged 6 to 16 years and 11 months. The WISC-III is divided into ten subtests (see Appendix), which are organized into verbal and performance scales. The subtests yield three composite scores: verbal IQ, performance IQ, and full-scale IQ - which estimate the individual's verbal language, nonverbal/visual-spatial/visual-motor, and general intellectual abilities, respectively.
Comparison of age, composite IQ scores, and standard subtests scores were performed by using the Kruskal-Wallis test, followed by the Dunn test for post-hoc analysis. Generalized Fisher's exact test was used for comparison of gender, and psychiatric symptoms and disorders among the syndromes. All analyses were performed using SAS software version 9.1.3 for Windows, with a significance level of 5%.
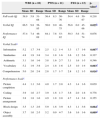
ResultsThe sample's sociodemographic characteristics are detailed in Table 1. Among the few participants who attended regular school (17%), only two (33%) completed elementary school. Table 2 presents the comparison among the three syndromes regarding age, composite IQ, and subtest scores. Frequencies of specific behaviors and psychiatric symptoms and disorders are displayed in Table 3.
Means, standard deviations, age ranges, and WISC-III subtests in WBS, PWS, and FXS.
| WBS (n=10) | PWS (n=11) | FXS (n=13) | p-valuea | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Range | Mean | SD | Range | Mean | SD | Range | ||
| Full-scale IQ | 58.9 | 5.9 | 51-66 | 56.4 | 8.3 | 50-76 | 54.4 | 4.6 | 50-67 | 0.218 |
| Verbal IQ | 66.5 | 9.8 | 56-85 | 56.0 | 9.0 | 46-75 | 56.8 | 6.0 | 45-66 | 0.025b |
| Performance IQ | 57.4 | 7.4 | 46-69 | 64.1 | 7.6 | 53-82 | 59.3 | 5.6 | 51-74 | 0.076 |
| Verbal Scalec | ||||||||||
| Information | 5.2 | 1.7 | 3-8 | 2.5 | 1.2 | 1-4 | 3.3 | 1.7 | 1-6 | 0.007d |
| Similarities | 4.8 | 1.9 | 3-8 | 3.4 | 1.8 | 1-6 | 3.6 | 1.2 | 2-6 | 0.337 |
| Arithmetic | 3.1 | 1.6 | 1-6 | 3.8 | 1.6 | 2-7 | 3.1 | 1.6 | 1-5 | 0.546 |
| Vocabulary | 5.2 | 1.9 | 3-8 | 2.4 | 1.8 | 1-6 | 2.5 | 1.0 | 1-4 | 0.003d |
| Comprehension | 5.0 | 2.0 | 2-8 | 2.8 | 1.7 | 1-7 | 2.8 | 1.2 | 1-5 | 0.016b |
| Performance Scalec | ||||||||||
| Picture completion | 4.4 | 1.1 | 3-6 | 4.0 | 1.7 | 2-8 | 4.3 | 1.4 | 1-6 | 0.639 |
| Coding | 3.9 | 1.6 | 2-7 | 3.9 | 1.6 | 1-7 | 3.6 | 2.0 | 1-8 | 0.778 |
| Picture arrangement | 3.8 | 1.8 | 1-6 | 4.9 | 1.8 | 2-8 | 4.7 | 1.4 | 3-8 | 0.355 |
| Block design | 3.5 | 1.3 | 2-6 | 5.9 | 1.6 | 3-9 | 4.3 | 1.1 | 3-6 | 0.004d |
| Object assembly | 3.7 | 1.0 | 2-5 | 5.2 | 0.9 | 4-7 | 3.6 | 1.6 | 1-6 | 0.009d |
FXS, Fragile X syndrome; PWS, Prader-Willi syndrome; WBS, Williams-Beuren syndrome; WISC-III, Wechsler Intelligence Scale for Children, third edition.
Significant values in bold.
Frequencies of psychiatric disorders/symptoms and behavioral features in WBS, PWS, and FXS.
| Frequency (%) | ||||
|---|---|---|---|---|
| WBS (n=10) | PWS (n=11) | FXS (n=13) | p-valuea | |
| Psychiatric Disorders | ||||
| Attention deficit hyperactivity disorder | 6 (60%) | 6 (54%) | 110 (77%) | 0.54 |
| Anxiety | 6 (60%) | 5 (45%) | 7 (53%) | 0.90 |
| Depression | 5 (50%) | 4 (36%) | 2 (15%) | 0.24 |
| Enuresis | 3 (30%) | 2 (18%) | 4 (30%) | 0.79 |
| Learning disability | 8 (80%) | 10 (91%) | 12 (92%) | 0.66 |
| Obsessive-compulsive disorder | 1 (10%) | 3 (27%) | 2 (15%) | 0.63 |
| Sleep disturbance | 5 (50%) | 8 (72%) | 4 (30%) | 0.12 |
| Symptoms and behaviors | ||||
| Aggression toward others | 3 (30%) | 6 (54%) | 7 (53%) | 0.51 |
| Explosiveness | 6 (60%) | 10 (91%) | 8 (61%) | 0.18 |
| Hyperactivity/Impulsivity | 6 (60%) | 7 (63%) | 12 (92%) | 0.14 |
| Hyperphagia | 4 (40%) | 9 (81%) | 3 (23%) | 0.01b |
| Inattention | 9 (90%) | 6 (54%) | 11 (84%) | 0.15 |
| Obsessions | 3 (30%) | 5 (45%) | 3 (23%) | 0.58 |
| Oppositional behavior | 5 (50%) | 9 (81%) | 6 (46%) | 0.17 |
| Phobias/fears | 6 (60%) | 2 (18%) | 4 (30%) | 0.15 |
| Self-injurious behaviors | 2 (20%) | 8 (72%) | 3 (23%) | 0.02b |
| Social interaction deficits | 2 (20%) | 5 (45%) | 9 (69%) | 0.07 |
FXS, Fragile X syndrome; PWS, Prader-Willi syndrome; WBS, Williams-Beuren syndrome.
Significant values in bold.
Significant differences were found among the three syndromes regarding verbal IQ and verbal and performance subtests (Table 2). Post-hoc analysis revealed that the WBS group presented significantly higher scores in relation to the PWS group concerning verbal IQ and information, vocabulary, and comprehension subtests (p < 0.05), and significantly higher scores in relation to the FXS group regarding vocabulary and comprehension subtests (p < 0.05). Additionally, the PWS group presented significantly higher scores in relation to the WBS and FXS groups concerning the block design and object assembly subtests (p < 0.05).
Results of the generalized Fisher's exact test demonstrated a significant difference among the three syndromes regarding frequencies of hyperphagia and self-injurious behaviors (Table 3).
DiscussionAlthough the present sample was relatively small, to the authors’ knowledge, this is the first study to specifically compare these three genetic syndromes using the same methodology of cognitive and behavior/psychiatric assessment in the developing world.
In this study, it was observed that children and adolescents with genetic syndromes and ID, who share equivalent degrees of intellectual impairment and come from similar social and economic contexts, exhibited a heterogeneous cognitive, behavioral, and psychopathological profile.
Predominance of language skills over visuospatial skills in children and adolescents with WBS was highlighted in some studies;8,13 however it was not identified in others.24,25 In the present study, good performance in verbal language activities (i.e., the ability to understand others and communicate appropriately) of children and adolescents with WBS became even more apparent when this syndrome was compared with FXS and PWS.
Another relevant result of this study was a significantly higher score obtained by children with PWS in visuospatial construction abilities (e.g., jigsaw puzzles, building models with bricks and pieces), which corroborates results from some previous studies5,9,26 and differs from others.27,28
These findings may help to raise the awareness of pediatricians and other healthcare professionals about the peculiar neuropsychomotor development and cognitive skills of individuals with these genetic syndromes, which could possibly lead to better informed rehabilitation efforts promoted by healthcare professionals.
Regarding the distinctive behavior profile among the three syndromes, Sarimski29 found a higher association between insatiable appetite and children with PWS; higher frequency of self-injurious behaviors, hyperactivity, aggression, and oppositional behavior in children with FXS; and higher prevalence of sleep disturbances and better social interaction in children with WBS. Di Nuovo and Buono30 reported lower communication skills in children with WBS compared to children with FXS.
In the present study, the comparison among children with WBS, PWS, and FXS regarding behavior features and psychiatrics symptoms/disorders revealed that the frequencies of hyperphagia and self-injurious behaviors were significantly higher in the PWS group than in the WBS and FSX groups.
Phobias and fears, inattention, and depression were more prevalent in WBS group. Children with PWS exhibited more oppositional behavior, explosiveness, sleep disturbance, obsessions, and obsessive-compulsive disorder. Hyperactivity and impulsivity, social interaction deficits, and attention deficit hyperactivity disorder were more frequent in the FXS group.
The differences in prevalence of psychiatric symptoms/disorders and specific behaviors between the syndromes justify a targeted care for these individuals. Pediatricians and other healthcare professionals should be familiar with the behavioral phenotype of different genetic syndromes with ID, tailoring pharmacological treatment and rehabilitation for each condition.
The study sample was relatively small, it was selected by convenience, and sample size calculation was not performed. Thus, the results achieved in this study should be regarded with caution regarding to their generalizability. Nevertheless, the data presented in this exploratory study are sufficiently robust to support the claim that these three syndromes have a distinctive cognitive and behavioral profile.
According to Salvador-Carulla and Bertelli,6 caring for patients with ID has been limited to the social and educational services. Thus, very little attention is paid by health professionals and scientists to this subject. In the case of genetic syndromes with relative low prevalence, such as WBS, PWS, and FXS, the knowledge gap on the part of healthcare professionals is even greater.
For the pediatricians and other health professionals, better understanding of the cognitive, behavioral, and psychopathological profiles of children and adolescents with genetic syndromes and with distinct forms of ID can inform the choice of the strategies for care and rehabilitation of these individuals. As a research topic, it may illuminate the complex relationship between genes, brain development, and expression of specific cognitive, behavioral and psychopathological features.
Conflicts of interestThe authors declare no conflits of interest.
The authors would like to thank the patients and their families for their participation.
Please cite this article as: Pegoraro LF, Steiner CE, Celeri EH, Banzato CE, Dalgalarrondo P. Cognitive and behavioral heterogeneity in genetic syndromes. J Pediatr (Rio J). 2014;90:155–60.





