

Hemoglobin SC is the second most common variant of sickle-cell disease worldwide, after hemoglobin SS. The objectives of the study were to describe the clinical and laboratory characteristics of hemoglobin SC disease in children from a newborn screening program and treated at a blood center.
MethodologyThis study assessed a cohort of 461 infants born between 01/01/1999 and 12/31/2012 and followed-up until 12/31/2014. Clinical events were expressed as rates for 100 patient-years, with 95% confidence intervals. Kaplan–Meier survival curves were created.
ResultsThe median age of patients was 9.2 years; 47.5% were female. Mean values of blood tests were: hemoglobin, 10.5g/dL; reticulocytes, 3.4%; white blood cells, 11.24×109/L; platelets, 337.1×109/L; and fetal hemoglobin, 6.3%. Clinical events: acute splenic sequestration in 14.8%, blood transfusion 23.4%, overt stroke in 0.2%. The incidence of painful vaso-occlusive episodes was 51 (48.9–53.4) per 100 patient-years and that of infections, 62.2 episodes (59.8–64.8) per 100 patient-years. Transcranial Doppler ultrasonography (n=71) was normal given the current reference values for SS patients. Hydroxyurea was given to ten children, all of whom improvement of painful crises. Retinopathy was observed in 20.3% of 59 children who underwent ophthalmoscopy. Avascular necrosis was detected in seven of 12 patients evaluated, predominantly in the left femur. Echocardiogram compatible with pulmonary hypertension was recorded in 4.6% of 130 children, with an estimated average systolic pulmonary artery pressure of 33.5mmHg. The mortality rate from all causes was 4.3%.
ConclusionsClinical severity is variable in SC hemoglobinopathy. Several children have severe manifestations similar to those with SS disease.
A hemoglobinopatia SC é a segunda variante mais comum da doença falciforme no mundo, após a hemoglobinopatia SS. Os objetivos do estudo foram descrever as características clínicas e laboratoriais da hemoglobinopatia SC em recém-nascidos diagnosticados por programa de triagem neonatal e encaminhados para acompanhamento em hemocentro.
MetodologiaCoorte de 461 recém-nascidos SC nascidos entre 01/01/1999 e 31/12/2012 e seguidos até 31/12/2014. A incidência de eventos clínicos foi expressa por taxas relativas a 100 pacientes-ano, com limites de confiança a 95%. Curvas de sobrevida foram construídas segundo Kaplan-Meier.
ResultadosMediana de idade, 9,2 anos; 47,5%, feminino. Médias dos valores hematológicos: hemoglobina 10,5g/dL; reticulócitos 3,4%; leucometria 11,24x109/L; plaquetometria 337,1x109/L; hemoglobina fetal 6,3%. Eventos clínicos: sequestro esplênico agudo em 14,8%, hemotransfusão 23,4%, AVC isquêmico 0,2%. A incidência de episódios vaso-oclusivos dolorosos foi de 51 (48,9-53,4) por 100 pacientes-ano; a de infecções, 62,2 episódios (59,8-64,8) por 100 pacientes-ano. Doppler transcraniano (n = 71) foi normal, se usados os valores de referência de crianças SS. Dez pacientes usaram hidroxiureia, todos com melhoria das crises dolorosas. Retinopatia foi observada em 20,3% das 59 crianças que fizeram fundoscopia. Necrose avascular foi detectada em 7 de 12 pacientes avaliados, com predomínio no fêmur esquerdo. Ecocardiograma compatível com hipertensão pulmonar foi registrado em 4,6% de 130 crianças, com média estimada de 33,5mm Hg de pressão arterial pulmonar. A taxa de mortalidade por todas as causas foi de 4,3%.
ConclusõesA hemoglobinopatia SC tem gravidade variável; várias crianças apresentam manifestações clínicas intensas, semelhantes às da hemoglobinopatia SS.
SC hemoglobinopathy is one of the most common variant of sickle-cell disease worldwide, second only to SS anemia. Hemoglobin C was first described in 1950 by Itano and Neel.1 It is caused by a mutation in the sixth codon of the beta globin gene, which results in the substitution of glutamic acid by lysine (GAG>AAG).
The process that leads to clinical complications in individuals with SC hemoglobinopathy is not yet fully understood and appears to be related more to hyperviscosity and cell dehydration than to hemolysis-related vasculopathy, characteristic of SS disease.2–5
The clinical events found in hemoglobin SS disease may also occur in patients with SC hemoglobinopathy.5,6 However, the clinical manifestations appear to be less severe, except for proliferative retinopathy and necrosis of the femoral head.2,5,6 Hydroxyurea seems to significantly reduce episodes of acute chest syndrome and painful vaso-occlusive crises (VOC) in children with SC hemoglobinopathy.2,5,7–9
The aim of this study was to describe the clinical and laboratory characteristics of hemoglobin SC disease in children from a neonatal screening program and followed at a blood center.
MethodsThe descriptive study was based on a retrospective cohort, using the medical records from the blood center and the neonatal screening database. The population initially consisted of 569 children with FSC electrophoretic profile at birth, who were born between 01/01/1999 and 12/31/2012, screened by the Neonatal Screening Program of Minas Gerais (PTN-MG), and followed-up at the Hemocentro of Belo Horizonte (HBH) of Fundação Hemominas.
Twelve children were excluded from the analysis due to transfer to another blood center and 18 due to loss of clinical follow-up; 539 individuals were still eligible. Specific contact with the families for signing the free and informed consent form (FICF) was not possible in 78 cases.
The clinical and laboratory findings refer to the subtotal of 461 children included in the study (539 minus the 78 whose families did not sign the FICF). The evolution of these children was reviewed up to 12/31/2014, so that all the children were followed-up for a minimum of two years.
Clinical information, such as VOC, infections, acute splenic sequestration crisis (ASSC), priapism, ophthalmologic, orthopedic and cardiologic consultations, and acute cerebrovascular events were extracted from the medical records, as well as data on treatment (blood transfusions and hydroxyurea use).
A transcranial Doppler (TCD) was carried out in 71 children and interpreted by a single professional using Nicolet equipment (model EME TC 2000, Nicolet – Madison, WI, USA). A high risk of ischemic stroke was defined as time-averaged mean of the maximum velocity (TAMMX)>200cm/s in the internal carotid or middle cerebral arteries, according to the STOP10 protocol.
A complete blood count was performed using the Coulter T-890 equipment (Beckman Coulter, Inc., CA, USA). The reticulocytes were counted in smear stained with brilliant cresyl blue. All hematological values were transcribed from the medical records, in the absence of infectious processes or painful crises, and at least three months after the use of blood components. The arithmetic mean of hematological data was considered as the baseline value for each patient. The baseline fetal hemoglobin (FHb) was obtained through hemoglobin electrophoresis at the most advanced age within the follow-up period, provided that it was collected after two years of life.
In order not to overestimate mortality, the 78 children who did not have a signed FICF were included in the rate denominator, since they were alive according to the medical follow-up records.
The statistical analysis was performed using SPSS (IBM SPSS Statistics for Windows, Version 20.0. NY, USA). The results were expressed as mean and standard deviation (SD), or median and interquartile range, when the distribution of values was non-Gaussian. The incidence of ASSC, VOC, infections, and blood transfusions were expressed by rates relative to 100 patient-years, with 95% confidence intervals. The probability curves for the occurrence of the first episode of ASSC and death (Kaplan–Meier) were extracted from the “One minus Survival function” chart, which represents the function (1 – Survival function).
The study was approved by the Research Ethics Committee of the institution, Opinion 13327713.5.0000.5149. The patients and parents/guardians were asked to sign the FICF.
ResultsOf the 461 children, 219 (47.5%) were girls. Age ranged from 1 to 16.7 years (median 9.2 years). At some point during the clinical course, 317 children (68.8%) had a palpable spleen below the left costal border, in the absence of an infectious process. The mean maximum values and the last spleen size value were 2.0 and 0.5cm below the costal border, respectively. The mean ages when these values were measured were 5.8 years and 8.6 years, respectively.
At least one VOC was recorded in 344 patients (74.6%), and 21.9% of the cases were hospitalized. The time to the first VOC ranged from 2 months to 15 years (median 5.1 years). The incidence of VOC was 51 crises per 100 patient-years (95% CI: 48.9–53.4). Approximately 25% of the children did not present VOC during the study period.
The incidence of infectious episodes was 62.2 per 100 patient-years (95% CI: 59.8–64.8). Only 36 children (7.8%) did not have any infection recorded in the medical files. Upper airway infections were the most frequent (26.5%), followed by pneumonia/acute chest syndrome (17.2%), tonsillitis (14.6%), acute otitis media (8%), and fever of unknown origin (6.2%).
ASSC was recorded in 68 patients (14.8%), with recurrence rate of 25%. The incidence was of 1.9 first events per 100 patient-years (95% CI: 1.5–2.4). The age at the first event ranged from 3.5 months to 12.8 years (median 3.8 years). Most of the first episodes of ASSC (70%) occurred within the first five years of life. The probability curve for the occurrence of the first ASSC episode is shown in Fig. 1. Seven children underwent splenectomy, two after the first episode, three after the second, and one after the third. The age at the procedure ranged from 2.8 to 12.7 years (median 5.7 years).
.")
Four boys (0.9%) had priapism episodes. One patient had two events in less than 30 days, aged 7.9 years, while the other three had one event each, at 6.3, 7.3, and 9.7 years of age.
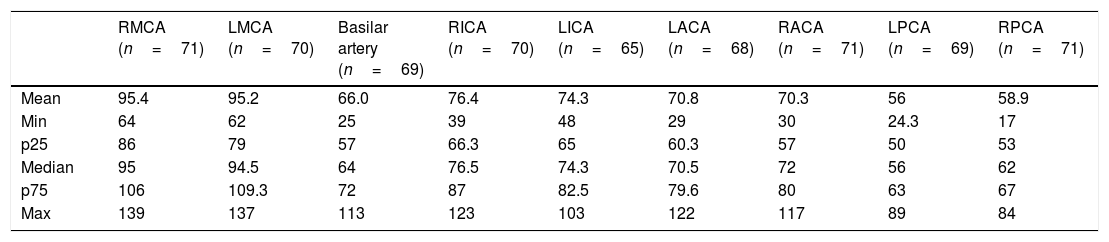
TCD was performed in 71 of the 461 children (15.4%), all with low risk of ischemic stroke (Table 1), using reference values for patients with SS hemoglobinopathy. One girl had ischemic stroke at age 11. Transthoracic and transesophageal echocardiographies were performed, which did not disclose intracardiac septal defects. The investigation of thrombophilia was negative. Cerebral magnetic resonance angiography evidenced bilateral vasculopathy, therefore being considered responsible for the cerebral ischemia. Sequencing of the HBB gene confirmed the S and C mutations and did not identify other mutations. The child had not undergone TCD before the stroke. At 15 years of age, TAMMX in the basilar artery was 113cm/s, suggesting compensatory circulation secondary to stroke. Currently, the child has an adequate social behavior, but shows mild to moderate claudication in the lower left limb and moderate deficit in mathematical reasoning. She receives regular blood transfusions and psychopedagogical support.
Distribution of transcranial Doppler maximum mean velocities (cm/s) in 71 children with SC hemoglobinopathy.
| RMCA (n=71) | LMCA (n=70) | Basilar artery (n=69) | RICA (n=70) | LICA (n=65) | LACA (n=68) | RACA (n=71) | LPCA (n=69) | RPCA (n=71) | |
|---|---|---|---|---|---|---|---|---|---|
| Mean | 95.4 | 95.2 | 66.0 | 76.4 | 74.3 | 70.8 | 70.3 | 56 | 58.9 |
| Min | 64 | 62 | 25 | 39 | 48 | 29 | 30 | 24.3 | 17 |
| p25 | 86 | 79 | 57 | 66.3 | 65 | 60.3 | 57 | 50 | 53 |
| Median | 95 | 94.5 | 64 | 76.5 | 74.3 | 70.5 | 72 | 56 | 62 |
| p75 | 106 | 109.3 | 72 | 87 | 82.5 | 79.6 | 80 | 63 | 67 |
| Max | 139 | 137 | 113 | 123 | 103 | 122 | 117 | 89 | 84 |
RMCA, right middle cerebral artery; LMCA, left middle cerebral artery; RICA, right internal carotid artery; LICA, left internal carotid artery; LACA, left anterior cerebral artery; RACA, right anterior cerebral artery; RPCA, right posterior cerebral artery; LPCA, left posterior cerebral artery.
A total of 232 children (50.3%) were referred to ophthalmologic assessment. Of this total, 59 (25.4%) underwent fundoscopy, and a finding compatible with retinopathy was verified in 20.3% of them. The median age at retinopathy diagnosis was 9.5 years. Regarding the orthopedic assessment, 12 children had a recorded medical consultation (2.6%), and avascular necrosis was observed in seven. Five showed a lesion in the left femur. One child had bilateral avascular necrosis in the shoulders and another had bilateral femoral lesions. As for the cardiologic assessment, 130 children (28.2%) underwent an echocardiogram during the follow-up period; 4.6% had findings suggestive of pulmonary hypertension, with an estimated mean of 33.5mmHg PASP, which varied between 31 and 38mmHg. No records of sickle cell nephropathy were retrieved from the files.
Regarding the clinical treatment, prescription of antimicrobial prophylaxis, folic acid supplementation, and immunization was recorded for all patients. It was not possible to adequately quantify treatment adherence, as there was no standardized recording of these data by the attending physicians.
Transfusion of packed red blood cells was necessary in 108 children (23.4%). As mentioned before, one patient received 63 transfusions as secondary ischemic stroke prevention. The other 107 patients received 185 transfusion procedures, with a mean incidence of 4.7 transfusions per 100 patient-years (95% CI: 4.0–5.4). They were caused by ASSC, preparation for surgical procedures, and clinical complications associated with infections. A total of 76.6% of the children did not receive transfusions; 15% received only one and 8.4%, two or more.
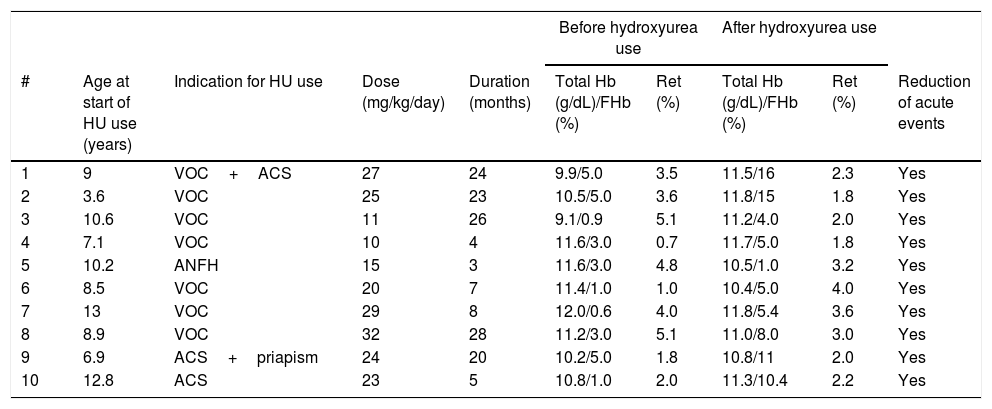
Ten (2.2%) children used hydroxyurea indicated due to repeated episodes of VOC in six cases. Avascular necrosis of the femoral head (one case) and ACS (three cases, alone or in combination with other events) comprised the other indications (Table 2). All had a significant reduction in the number of VOC. The child with bone necrosis showed marked gait improvement and had significant pain reduction.
Clinical and laboratory data of ten children with SC hemoglobinopathy who used hydroxyurea (HU).
| Before hydroxyurea use | After hydroxyurea use | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | Age at start of HU use (years) | Indication for HU use | Dose (mg/kg/day) | Duration (months) | Total Hb (g/dL)/FHb (%) | Ret (%) | Total Hb (g/dL)/FHb (%) | Ret (%) | Reduction of acute events |
| 1 | 9 | VOC+ACS | 27 | 24 | 9.9/5.0 | 3.5 | 11.5/16 | 2.3 | Yes |
| 2 | 3.6 | VOC | 25 | 23 | 10.5/5.0 | 3.6 | 11.8/15 | 1.8 | Yes |
| 3 | 10.6 | VOC | 11 | 26 | 9.1/0.9 | 5.1 | 11.2/4.0 | 2.0 | Yes |
| 4 | 7.1 | VOC | 10 | 4 | 11.6/3.0 | 0.7 | 11.7/5.0 | 1.8 | Yes |
| 5 | 10.2 | ANFH | 15 | 3 | 11.6/3.0 | 4.8 | 10.5/1.0 | 3.2 | Yes |
| 6 | 8.5 | VOC | 20 | 7 | 11.4/1.0 | 1.0 | 10.4/5.0 | 4.0 | Yes |
| 7 | 13 | VOC | 29 | 8 | 12.0/0.6 | 4.0 | 11.8/5.4 | 3.6 | Yes |
| 8 | 8.9 | VOC | 32 | 28 | 11.2/3.0 | 5.1 | 11.0/8.0 | 3.0 | Yes |
| 9 | 6.9 | ACS+priapism | 24 | 20 | 10.2/5.0 | 1.8 | 10.8/11 | 2.0 | Yes |
| 10 | 12.8 | ACS | 23 | 5 | 10.8/1.0 | 2.0 | 11.3/10.4 | 2.2 | Yes |
HU, hydroxyurea; VOC, painful vaso-occlusive crises; Ret, reticulocytes; ACS, acute chest syndrome; ANFH, avascular necrosis of the femoral head.
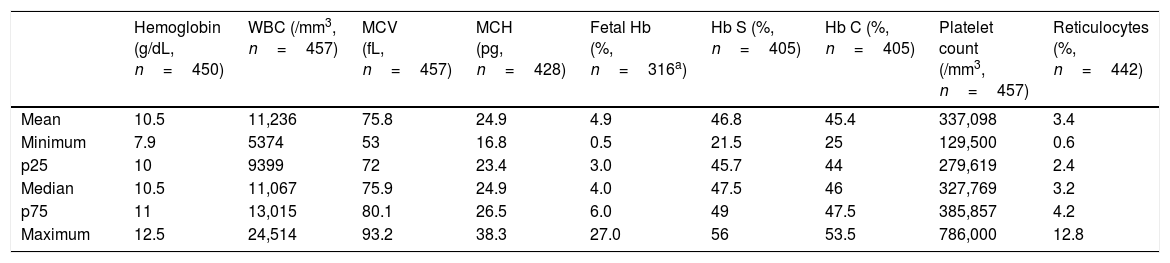
The mean hemoglobin was 10.5g/dL and the mean reticulocyte count was 3.4%. The distribution of hematological exams is shown in a summarized form in Table 3.
Hematologic exams in children with SC hemoglobinopathy treated in a Brazilian state Blood Center.
| Hemoglobin (g/dL, n=450) | WBC (/mm3, n=457) | MCV (fL, n=457) | MCH (pg, n=428) | Fetal Hb (%, n=316a) | Hb S (%, n=405) | Hb C (%, n=405) | Platelet count (/mm3, n=457) | Reticulocytes (%, n=442) | |
|---|---|---|---|---|---|---|---|---|---|
| Mean | 10.5 | 11,236 | 75.8 | 24.9 | 4.9 | 46.8 | 45.4 | 337,098 | 3.4 |
| Minimum | 7.9 | 5374 | 53 | 16.8 | 0.5 | 21.5 | 25 | 129,500 | 0.6 |
| p25 | 10 | 9399 | 72 | 23.4 | 3.0 | 45.7 | 44 | 279,619 | 2.4 |
| Median | 10.5 | 11,067 | 75.9 | 24.9 | 4.0 | 47.5 | 46 | 327,769 | 3.2 |
| p75 | 11 | 13,015 | 80.1 | 26.5 | 6.0 | 49 | 47.5 | 385,857 | 4.2 |
| Maximum | 12.5 | 24,514 | 93.2 | 38.3 | 27.0 | 56 | 53.5 | 786,000 | 12.8 |
Among the 539 children initially enrolled for the study, there were 23 deaths (4.3%). In six cases it was not possible to determine the cause of death. Ten deaths were related to infectious complications, with six episodes of pneumonia/ACS and four episodes of sepsis. Of the seven remaining deaths, three occurred after ASSC. Acute aplastic crisis, gastroenteritis/dehydration, traumatic brain injury, and hemorrhagic shock were responsible for one death each. The age at death ranged from 3 months to 9.7 years, with a median of 3.2 years; 65.2% died younger than 5 years of age. The estimated probability of death until the age of 10 years was 5.4% (95% CI: 3.2–7.6%).
DiscussionThe present study included 461 children with SC hemoglobinopathy. The severity of the clinical manifestations in SC disease varied from child to child. As shown by the maximum incidence rates of the different clinical events, several children had high values, suggesting a severity of the clinical course similar to that observed in SS children.
Approximately 75% of the patients had at least one VOC, a result similar to the 78% found by Tuttle and Koch,11 but superior to that reported by Powars et al.,12 with 60% of cases. The incidence of VOC for the entire population in the present study was 51 crises per 100 patient-years. The American cohort had a mean incidence of 30 VOC per 100 patient-year.13 The same group observed that the time until the first report of pain in childhood was influenced by the disease genotype, occurring significantly earlier in SS disease (median of 13.9 months) than in SC disease (median of 43.6 months).14 In the present study, the median was 61 months. Painful crises in children with SC hemoglobinopathy occur with approximately half the frequency of what is observed in sickle-cell anemia.2,5,12,13 However, it should be emphasized that even though the incidence is lower, it is relevant and should be considered by healthcare teams in emergency units, in order to avoid under-treatment and dismissal of pain complaints in patients with SC hemoglobinopathy.
Regarding the evaluation of target organs, a finding compatible with retinopathy was observed in 20.3% children submitted to fundoscopy. Most studies on retinopathy have been performed in adult patients, with a mean prevalence of approximately 33%.2,3,15 Among the studies that included children, the result was similar to that found by two North American groups, with 23% and 16.4%, respectively.13,16 Regarding the cardiological evaluation, 4.6% of the children who underwent echocardiography presented echocardiographic values that were suggestive of pulmonary hypertension, a result similar to that found in an English study,17 with 6%. However, what is noteworthy in the present study was the small number of children submitted to fundoscopy (25%) and echocardiogram (28.2%), since patients should be referred for annual ophthalmologic and cardiologic evaluations after the age of 5 years. It is possible that the actual incidence of sickle cell retinopathy and echocardiographic alterations suggestive of pulmonary hypertension are underestimated in the assessed population. This demonstrates the need to establish a permanent network of experienced interconsultants in proliferative retinopathy and pediatric cardiology for adequate early detection and treatment.
Regarding the orthopedic evaluation, a description of avascular necrosis was found in 58.3% of the assessed patients. In the literature, the prevalence varies between 8.8 and 15%,3,12,13,18 but the small number of cases in the present study hinders the establishment of a solid conclusion. Four boys (0.9%) presented priapism episodes during the follow-up period. Published data on priapism usually result from the joint analysis of the SS, SC, and S-beta thalassemia genotypes, with an approximate prevalence of 35% in adults.19 Likewise, the small number of cases and the retrospective design of the study limit the data analysis.
Sixty-eight children (14.8%) had ASSC. There are few studies on ASSC involving children with SC hemoglobinopathy; the prevalence is estimated to be between 5 and 12%.4,20,21 Most of the ASSC early episodes in the present study (70%) occurred within the first five years of life. A study performed by the same authors on 255 SS/β0 thalassemia children treated at the Belo Horizonte Blood Center showed that, in addition to the incidence of ASSC being approximately five times higher in children with SS/Sβ0 thalassemia (10.2 first episodes per 100 patient-years), 75% of them presented the first event before the age of 2 years.22 There was a convergence in the indication of splenectomy after two or more ASSC episodes in both studies. Most authors, however, recommend performing the surgical procedure after the first or, at most, after the second episode of ASSC.18,21 The authors believe that, considering the presented data, the education of caregivers of children with SC disease in relation to spleen palpation is, therefore, as important as in children with SS disease.
Regarding baseline hematological laboratory tests (Table 3), the mean hemoglobin levels, white blood cells, reticulocyte counts and platelet counts were 10.5g/dL, 11,236/mm3, 3.4% and 337.098/mm3, respectively. Anemia in SC hemoglobinopathy is generally milder than in SS patients, with mean hemoglobin values ranging from 10.5–11.5g/dL.3,23 As previously described, reticulocytes are slightly increased, and platelet and leukocyte counts are within the normal range.2,5
All children who underwent TCD showed a low risk of ischemic stroke (Table 1). It is known that the incidence of stroke is approximately four times lower than that in patients with SS hemoglobinopathy, with rates ranging between 0.8 and 3%.12,24 There are currently insufficient studies and data regarding the TAMMX reference values for patients with SC hemoglobinopathy, and the classification of patients with SS hemoglobinopathy is used in these cases.18,24,25 Vieira et al.26 reported that 0.7% of exams presented a high risk of stroke in 558 children with SC hemoglobinopathy. These authors suggest that lower reference values should be used in children with SC to be characterized as at high risk for ischemic stroke, but this requires further studies.
One child in the present study had an ischemic stroke episode. Deane et al. described the case of a patient with SC hemoglobinopathy who had an ischemic stroke at the age of 5 years, considered secondary to the underlying disease after ruling out other possibilities; the TAMMX was 146cm/s in the right middle cerebral artery and 126cm/s in the left middle cerebral artery.27 A case of stroke in a 7-year-old child was also described by Powars et al.28 in 1978 and another in a 6-year-old Nigerian child by Lagunju and Brown29 in 2012. Three pediatric cases were reported by Ohene-Frempong in a North-American collaboration study.24 As previously mentioned, there are no current data on TAMMX values to perform stratify ischemic stroke risk in patients with SC hemoglobinopathy. Therefore, the TAMMX results of the present study (Table 1) contribute to fill some gaps in the understanding of cerebral vasculopathy of SC children.
The use of hydroxyurea caused significant symptom reduction, similar to what has been previously described.7–9 It was observed that no patient received the maximum dose of hydroxyurea (35mg/kg/day) due to the supervenience of hematological toxicity. Luchtman-Jones et al. reported 133 children with SC hemoglobinopathy treated with hydroxyurea at 18 referral centers in the United States. Their study showed a 38% reduction in painful events after 12 months of drug use.9
The estimated probability of death at 10 years, from all causes, was 5.4%, well below the 13.7% rate recorded for children with SS/Sβ0 thalassemia and similar to the 4.4% recorded in a study that included the entire state of Minas Gerais, Brazil.30 The United States collaboration group reported a 9% mortality rate in a cohort of 284 patients.13 It is observed that most of the children (65.2%) died when aged younger than 5 years. This finding can be explained by the higher incidence, in the described age group, of severe acute events and high potential for lethality, such as infections and ASSC, the most frequent causes of death in the present study. A significant occurrence of deaths secondary to indeterminate causes (26%) was also observed, which corroborates the difficulties of health teams in identifying possible clinical complications secondary to sickle cell disease.
The main limitations of the present study include the retrospective analysis and the small amount of data on specialized consultations of the patients included in the study, which restricts the conclusions about the incidence of some clinical complications, such as proliferative retinopathy and necrosis of the head of long bones. The evaluation of the hydroxyurea effects also has sampling limitations. The main reason for the sample loss of 19% (108/569) in the clinical follow-up refers to the difficulty in collecting the signed FICF, which accounted for 72% of the losses. The analysis of epidemiological data such as gender, age, and number of visits recorded in the Blood Center charts does not indicate that these children would have a clinical course different from that of the 461 children included in the study, but this possibility cannot be ruled out. Although only children from the state of Minas Gerais were included in this study, it is possible that the results shown herein may be extrapolated to other states, since the C allele has a unicentric origin,2 although other genetic and environmental factors may have a different effect on children from other states and countries.
In conclusion, the analysis of the results adds relevant information about the profile of the clinical and laboratory behavior of SC hemoglobin disease in Brazil, aiming at preventive and curative health actions. Acute complications and target-organ lesions occur at varying degrees and several children show intense clinical manifestations, similar to those observed in SS hemoglobinopathy.
FundingFundação Hemominas, Newborn Screening Program (Nupad-UFMG), Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflicts of interestThe authors declare no conflicts of interest.
To CNPq, Fapemig, and Nupad financial support to the project. To our colleagues from Fundação Hemominas for the logistical support. To the children and their families for the consent to perform the research and publication of the results.
Please cite this article as: Rezende PV, Santos MV, Campos GF, Vieira LL, Souza MB, Belisário AR, et al. Clinical and hematological profile in a newborn cohort with hemoglobin SC. J Pediatr (Rio J). 2018;94:666–72.




