

Hemoglobin SC is the second most common variant of sickle‐cell disease worldwide, after hemoglobin SS. The objectives of the study were to describe the clinical and laboratory characteristics of hemoglobin SC disease in children from a newborn screening program and treated at a blood center.
MethodologyThis study assessed a cohort of 461 infants born between 01/01/1999 and 12/31/2012 and followed‐up until 12/31/2014. Clinical events were expressed as rates for 100 patient‐years, with 95% confidence intervals. Kaplan–Meier survival curves were created.
ResultsThe median age of patients was 9.2 years; 47.5% were female. Mean values of blood tests were: hemoglobin, 10.5g/dL; reticulocytes, 3.4%; white blood cells, 11.24×109/L; platelets, 337.1×109/L; and fetal hemoglobin, 6.3%. Clinical events: acute splenic sequestration in 14.8%, blood transfusion 23.4%, overt stroke in 0.2%. The incidence of painful vaso‐occlusive episodes was 51 (48.9–53.4) per 100 patient‐years and that of infections, 62.2 episodes (59.8–64.8) per 100 patient‐years. Transcranial Doppler ultrasonography (n=71) was normal given the current reference values for SS patients. Hydroxyurea was given to ten children, all of whom improvement of painful crises. Retinopathy was observed in 20.3% of 59 children who underwent ophthalmoscopy. Avascular necrosis was detected in seven of 12 patients evaluated, predominantly in the left femur. Echocardiogram compatible with pulmonary hypertension was recorded in 4.6% of 130 children, with an estimated average systolic pulmonary artery pressure of 33.5mmHg. The mortality rate from all causes was 4.3%.
ConclusionsClinical severity is variable in SC hemoglobinopathy. Several children have severe manifestations similar to those with SS disease.
A hemoglobinopatia SC é a segunda variante mais comum da doença falciforme no mundo, após a hemoglobinopatia SS. Os objetivos do estudo foram descrever as características clínicas e laboratoriais da hemoglobinopatia SC em recém‐nascidos diagnosticados por programa de triagem neonatal e encaminhados para acompanhamento em hemocentro.
MetodologiaCoorte de 461 recém‐nascidos SC nascidos entre 01/01/1999 e 31/12/2012 e seguidos até 31/12/2014. A incidência de eventos clínicos foi expressa por taxas relativas a 100 pacientes‐ano, com limites de confiança a 95%. Curvas de sobrevida foram construídas segundo Kaplan‐Meier.
ResultadosMediana de idade, 9,2 anos; 47,5%, feminino. Médias dos valores hematológicos: hemoglobina 10,5g/dL; reticulócitos 3,4%; leucometria 11,24 x 109/L; plaquetometria 337,1x109/L; hemoglobina fetal 6,3%. Eventos clínicos: sequestro esplênico agudo em 14,8%, hemotransfusão 23,4%, AVC isquêmico 0,2%. A incidência de episódios vaso‐oclusivos dolorosos foi de 51 (48,9‐53,4) por 100 pacientes‐ano; a de infecções, 62,2 episódios (59,8‐64,8) por 100 pacientes‐ano. Doppler transcraniano (n = 71) foi normal, se usados os valores de referência de crianças SS. Dez pacientes usaram hidroxiureia, todos com melhoria das crises dolorosas. Retinopatia foi observada em 20,3% das 59 crianças que fizeram fundoscopia. Necrose avascular foi detectada em 7 de 12 pacientes avaliados, com predomínio no fêmur esquerdo. Ecocardiograma compatível com hipertensão pulmonar foi registrado em 4,6% de 130 crianças, com média estimada de 33,5mm Hg de pressão arterial pulmonar. A taxa de mortalidade por todas as causas foi de 4,3%.
ConclusõesA hemoglobinopatia SC tem gravidade variável; várias crianças apresentam manifestações clínicas intensas, semelhantes às da hemoglobinopatia SS.
A hemoglobinopatia SC é a segunda variante mais comum da doença falciforme no mundo, logo depois da anemia SS. A hemoglobina C foi descrita inicialmente em 1950 por Itano & Neel.1 Decorre de mutac¿a¿o no sexto códon do gene da beta globina, o que resulta na substituição de ácido glutâmico por lisina (GAG>AAG).
O processo que levaria às complicações clínicas em indivíduos com hemoglobinopatia SC não é ainda completamente compreendido e parece estar mais relacionado com hiperviscosidade e desidratação celular do que com a vasculopatia relacionada à hemólise, característica da doença SS.2–5
Os eventos clínicos encontrados na doença da hemoglobina SS podem também ocorrer em pacientes com hemoglobinopatia SC.5,6 Contudo, a gravidade das manifestações clínicas parece ser menos intensa, exceto quanto à¿ retinopatia proliferativa e necrose da cabeça do fêmur.2,5,6 A hidroxiureia parece reduzir significativamente os episódios de síndrome torácica aguda e crises vaso‐oclusivas dolorosas (CVO) em crianças com hemoglobinopatia SC.2,5,7–9
O objetivo deste estudo foi descrever as características clínicas e laboratoriais da doença SC em crianças provenientes de programa de triagem neonatal e seguidas num hemocentro.
MétodosO estudo descritivo baseou‐se em coorte retrospectiva, usaram‐se os prontuários médicos arquivados em hemocentro e do banco de dados da triagem neonatal. A população consistiu, inicialmente, de 569 crianças com perfil eletroforético FSC ao nascimento, entre 01/01/1999 e 31/12/2012, triadas pelo Programa de Triagem Neonatal de Minas Gerais (PTN‐MG) e acompanhadas no Hemocentro de Belo Horizonte (HBH) da Fundaça¿o Hemominas.
Foram excluídas da análise 12 crianças devido à transferência para outro hemocentro e 18 por perda de seguimento clínico, restaram 539 indivíduos. Contato específico com as famílias para assinatura do termo de consentimento livre e esclarecido (TCLE) não foi possível em 78 casos.
Os resultados clínicos e laboratoriais referem‐se ao subtotal de 461 crianças com TCLE (539 menos 78). Reviu‐se a evolução dessas crianças até 31/12/2014, de modo que todas as crianças foram acompanhadas pelo mínimo de dois anos.
Foram extraídas dos prontuários médicos informações clínicas, como CVO, infecções, sequestração esplênica aguda (SEA), priapismo, consultas oftalmológicas, ortopédicas e cardiológicas e eventos cerebrais vasculares agudos, bem como sobre o tratamento (hemotransfusões e uso de hidroxiureia).
Doppler transcraniano (DTC) foi feito em 71 crianças e interpretado por um único profissional, com equipamento Nicolet (modelo EME TC 2000, Nicolet, Madison, WI, EUA). Alto risco de acidente vascular cerebral isquêmico (AVCi) foi definido como velocidade média máxima (VMMax)>200cm/s nas artérias carótida interna ou cerebral média, conforme protocolo STOP.10
Fez‐se hemograma completo com o equipamento Coulter T‐890 (Beckman Coulter, Inc., CA, EUA). Os reticulócitos foram contados em esfregaço corado com azul de cresil brilhante. Todos os valores hematológicos foram transcritos dos prontuários, na ausência de processos infecciosos ou de crise álgica e, no mínimo, três meses após o uso de hemocomponentes. Considerou‐se a média aritmética dos dados hematológicos como valor basal para cada paciente. Pela eletroforese de hemoglobina obteve‐se o valor basal de hemoglobina fetal (HbF) na idade mais avançada dentro do período de acompanhamento, desde que colhida após os dois anos de vida.
Para que a mortalidade não ficasse superestimada, as 78 crianças sem assinatura de TCLE foram incluídas no denominador da taxa, pois se encontravam vivas segundo os prontuários médicos de acompanhamento.
A análise estati¿stica foi feita pelo programa SPSS (IBM SPSS Statistics for Windows, versão 20.0. NY, EUA). Os resultados foram expressos pela média e desvio‐padra¿o (DP), ou pela mediana e intervalo interquartil, quando a distribuic¿a¿o de valores era na¿o gaussiana. As incidências de SEA, CVO, infecções e transfusões sanguíneas foram expressas por taxas relativas a 100 pacientes‐ano, com limites de confiança a 95%. As curvas de probabilidade para a ocorrência do primeiro episódio de SEA e de óbito (Kaplan‐Meier) foram extraídas do gráfico One Minus Survival, que representa a função (1 – Função de sobrevida).
O estudo foi aprovado pelo Comitê de Ética em Pesquisa da instituição, parecer 13327713.5.0000.5149. Solicitou‐se assinatura do TCLE aos pacientes e responsáveis.
ResultadosDas 461 crianc¿as, 219 (47,5%) eram meninas. A idade variou de um a 16,7 anos (mediana 9,2 anos). Em algum momento da evolução clínica, 317 crianças (68,8%) apresentavam baço palpável abaixo do rebordo costal esquerdo, na ausência de processo infeccioso. As médias do valor máximo e do último valor do tamanho do baço foram de 2,0 e 0,5 centímetros abaixo do rebordo costal, respectivamente. As médias de idade quando da medida desses valores foram 5,8 anos e 8,6 anos, respectivamente.
Pelo menos uma CVO foi registrada em 344 pacientes (74,6%), houve hospitalização em 21,9%. O tempo para a primeira CVO variou entre dois meses e 15 anos (mediana 5,1 anos). A incidência de CVO foi de 51 crises por 100 pacientes‐ano (IC 95%: 48,9‐53,4). Aproximadamente 25% das crianças não tiveram CVO registrada durante o período de observação.
A incidência de episódios infecciosos foi de 62,2 por 100 pacientes‐ano (IC 95%: 59,8‐64,8). Apenas 36 crianças (7,8%) não apresentaram infecção registrada no prontuário médico. Infecção de vias aéreas superiores foi a mais frequente (26,5%), seguida de pneumonia/síndrome torácica aguda (17,2%), amigdalite (14,6%), otite média aguda (8%) e febre sem foco definido (6,2%).
SEA foi registrado em 68 pacientes (14,8%), com recorrência de 25%. A incidência foi de 1,9 primeiro evento por 100 pacientes‐ano (IC 95%: 1,5‐2,4). A idade ao primeiro evento variou entre 3,5 meses e 12,8 anos (mediana 3,8 anos). A maior parte dos primeiros episódios de SEA (70%) ocorreu até cinco anos de vida. A curva de probabilidade de ocorrência do primeiro episódio de SEA está representada na figura 1. Sete crianças foram submetidas a esplenectomia, duas após o primeiro episódio, três após o segundo e uma após o terceiro. A idade de feitura do procedimento variou entre 2,8 e 12,7 anos (mediana 5,7 anos).
.")
Quatro meninos (0,9%) tiveram episódios de priapismo. Um paciente teve dois eventos em intervalo inferior a 30 dias, com 7,9 anos, enquanto os outros três tiveram um evento cada, com 6,3, 7,3 e 9,7 anos.
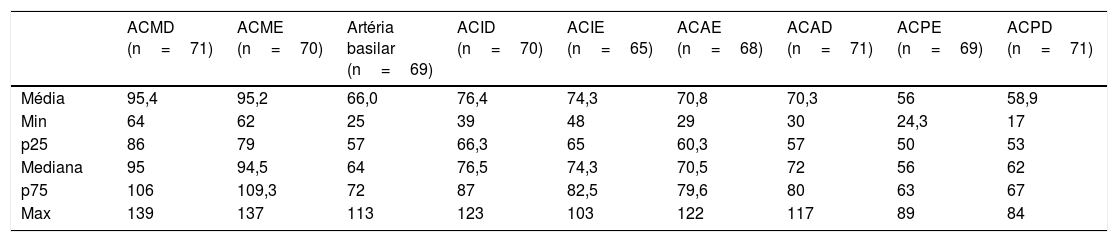
DTC foi feito em 71 das 461 crianças (15,4%), todas com baixo risco de AVCi (tabela 1), usados os valores de referência para pacientes com hemoglobinopatia SS. Uma menina apresentou AVCi aos 11 anos. Foram feitas ecocardiografias transtorácica e transesofágica que não revelaram comunicações intracardíacas. A investigação de trombofilias foi negativa. Angiorressonância cerebral evidenciou vasculopatia bilateral, foi considerada, portanto, responsável pela isquemia cerebral. Sequenciamento do gene HBB confirmou as mutações S e C e nenhuma outra mutação. A criança não havia feito DTC antes do AVCi. Aos 15 anos, a VMMax em artéria basilar era 113cm/seg, sugeriu circulação compensatória secundária ao AVCi. Atualmente, a criança apresenta convívio social adequado, porém tem leve a moderada claudicação no membro inferior esquerdo e moderada lentidão no raciocínio matemático. Encontra‐se em hemotransfusão regular e acompanhamento psicopedagógico.
Distribuição das velocidades médias máximas (cm/seg) do Doppler transcraniano em 71 crianças com hemoglobinopatia SC
| ACMD (n=71) | ACME (n=70) | Artéria basilar (n=69) | ACID (n=70) | ACIE (n=65) | ACAE (n=68) | ACAD (n=71) | ACPE (n=69) | ACPD (n=71) | |
|---|---|---|---|---|---|---|---|---|---|
| Média | 95,4 | 95,2 | 66,0 | 76,4 | 74,3 | 70,8 | 70,3 | 56 | 58,9 |
| Min | 64 | 62 | 25 | 39 | 48 | 29 | 30 | 24,3 | 17 |
| p25 | 86 | 79 | 57 | 66,3 | 65 | 60,3 | 57 | 50 | 53 |
| Mediana | 95 | 94,5 | 64 | 76,5 | 74,3 | 70,5 | 72 | 56 | 62 |
| p75 | 106 | 109,3 | 72 | 87 | 82,5 | 79,6 | 80 | 63 | 67 |
| Max | 139 | 137 | 113 | 123 | 103 | 122 | 117 | 89 | 84 |
ACAD, artéria cerebral anterior direita; ACAE, artéria cerebral anterior esquerda; ACID, artéria carótida interna direita; ACIE, artéria carótida interna esquerda; ACMD, artéria cerebral média direita; ACME, artéria cerebral média esquerda; ACPD, artéria cerebral posterior direita; ACPE, artéria cerebral posterior esquerda.
Foram encaminhadas para avaliação oftalmológica 232 crianças (50,3%). Desse total, foi registrada fundoscopia em 59 (25,4%), foi verificada anotação compatível com retinopatia em 20,3% delas. A idade mediana da identificação da retinopatia foi de 9,5 anos. Quanto à avaliação ortopédica, foi encontrado registro de consulta em 12 crianças (2,6%), com descrição de necrose avascular em sete. Em cinco, havia lesão do fêmur esquerdo. Uma criança apresentou necrose avascular bilateral nos ombros e outra apresentou lesão bilateral em fêmur. Quanto à avaliação cardiológica, 130 crianças (28,2%) submeteram‐se a ecocardiograma durante o período de seguimento; 4,6% apresentaram exame sugestivo de hipertensão pulmonar, com média estimada de 33,5mmHg de PSAP, que variou entre 31 e 38mmHg. Não foi encontrado nos prontuários médicos registro de nefropatia falciforme.
Em relação ao tratamento clínico, a prescrição de profilaxia antimicrobiana, suplementação com ácido fólico e imunização ocorreram para todos os pacientes. Não foi possível quantificar adequadamente a adesão ao tratamento, já que não houve padronização no registro desses dados pelos médicos assistentes.
Transfusão de concentrado de hemácias foi necessária em 108 crianças (23,4%). Uma paciente recebeu 63 transfusões como forma de prevenção secundária de AVCi, conforme já assinalado. As demais 107 receberam 185 procedimentos transfusionais, com incidência média de 4,7 transfusões por 100 pacientes‐ano (IC 95%: 4,0‐5,4). Foram motivadas por SEA, preparo para procedimentos cirúrgicos e intercorrências clínicas associadas a infecções. Não receberam transfusão 76,6% das crianças; 15% receberam uma única e 8,4%, duas ou mais.
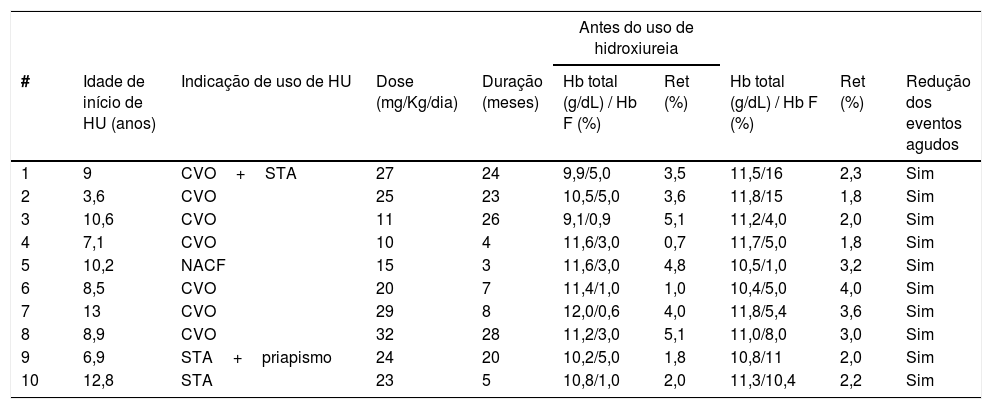
Dez (2,2%) crianças encontram‐se em uso de hidroxiureia indicada por CVO de repetição em seis casos. Necrose avascular de cabeça de fêmur (um caso) e STA (três casos, isoladamente ou em combinação com outros eventos) constituíram as outras indicações (tabela 2). Todos apresentaram significativa redução das CVOs. A criança com necrose óssea evoluiu com melhoria acentuada da marcha e expressiva redução da dor.
Dados clínicos e laboratoriais em 10 crianças com hemoglobinopatia SC que usaram hidroxiureia (HU)
| Antes do uso de hidroxiureia | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | Idade de início de HU (anos) | Indicação de uso de HU | Dose (mg/Kg/dia) | Duração (meses) | Hb total (g/dL) / Hb F (%) | Ret (%) | Hb total (g/dL) / Hb F (%) | Ret (%) | Redução dos eventos agudos |
| 1 | 9 | CVO+STA | 27 | 24 | 9,9/5,0 | 3,5 | 11,5/16 | 2,3 | Sim |
| 2 | 3,6 | CVO | 25 | 23 | 10,5/5,0 | 3,6 | 11,8/15 | 1,8 | Sim |
| 3 | 10,6 | CVO | 11 | 26 | 9,1/0,9 | 5,1 | 11,2/4,0 | 2,0 | Sim |
| 4 | 7,1 | CVO | 10 | 4 | 11,6/3,0 | 0,7 | 11,7/5,0 | 1,8 | Sim |
| 5 | 10,2 | NACF | 15 | 3 | 11,6/3,0 | 4,8 | 10,5/1,0 | 3,2 | Sim |
| 6 | 8,5 | CVO | 20 | 7 | 11,4/1,0 | 1,0 | 10,4/5,0 | 4,0 | Sim |
| 7 | 13 | CVO | 29 | 8 | 12,0/0,6 | 4,0 | 11,8/5,4 | 3,6 | Sim |
| 8 | 8,9 | CVO | 32 | 28 | 11,2/3,0 | 5,1 | 11,0/8,0 | 3,0 | Sim |
| 9 | 6,9 | STA+priapismo | 24 | 20 | 10,2/5,0 | 1,8 | 10,8/11 | 2,0 | Sim |
| 10 | 12,8 | STA | 23 | 5 | 10,8/1,0 | 2,0 | 11,3/10,4 | 2,2 | Sim |
CVO, crises vaso‐oclusivas dolorosas; HU, hidroxiureia; NACF, necrose avascular de cabeça de fêmur; Ret, reticulócitos; STA, síndrome torácica aguda.
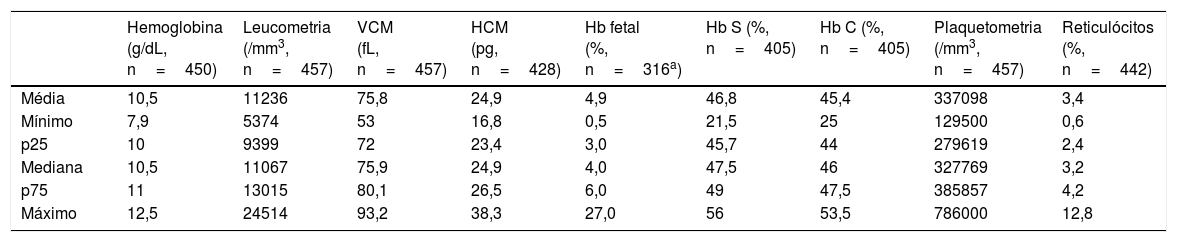
A hemoglobina média foi de 10,5g/dL e a contagem média de reticulócitos, 3,4%. A distribuição de valores dos exames hematológicos está representada, de forma condensada, na tabela 3.
Exames hematológicos em crianças com hemoglobinopatia SC atendidas em hemocentro de estado brasileiro
| Hemoglobina (g/dL, n=450) | Leucometria (/mm3, n=457) | VCM (fL, n=457) | HCM (pg, n=428) | Hb fetal (%, n=316a) | Hb S (%, n=405) | Hb C (%, n=405) | Plaquetometria (/mm3, n=457) | Reticulócitos (%, n=442) | |
|---|---|---|---|---|---|---|---|---|---|
| Média | 10,5 | 11236 | 75,8 | 24,9 | 4,9 | 46,8 | 45,4 | 337098 | 3,4 |
| Mínimo | 7,9 | 5374 | 53 | 16,8 | 0,5 | 21,5 | 25 | 129500 | 0,6 |
| p25 | 10 | 9399 | 72 | 23,4 | 3,0 | 45,7 | 44 | 279619 | 2,4 |
| Mediana | 10,5 | 11067 | 75,9 | 24,9 | 4,0 | 47,5 | 46 | 327769 | 3,2 |
| p75 | 11 | 13015 | 80,1 | 26,5 | 6,0 | 49 | 47,5 | 385857 | 4,2 |
| Máximo | 12,5 | 24514 | 93,2 | 38,3 | 27,0 | 56 | 53,5 | 786000 | 12,8 |
HCM, hemoglobina corpuscular média; VCM, volume corpuscular médio.
Entre as 539 crianças inicialmente arroladas para o estudo, houve 23 óbitos (4,3%). Em seis casos não foi possível determinar a causa do óbito. Houve dez óbitos relacionados a complicações infecciosas, foram seis episódios de pneumonia/síndrome torácica aguda (STA) e quatro de sepse. Dos sete óbitos remanescentes, três ocorreram após SEA. Crise aplástica aguda, gastroenterite/desidratação, traumatismo crânio‐encefálico e choque hemorrágico foram responsáveis por um óbito. A idade ao óbito variou entre três meses e 9,7 anos, com mediana de 3,2 anos; 65,2% faleceram com idade inferior a cinco anos. A probabilidade estimada de óbito até os dez anos de vida foi de 5,4% (I.C. 95%: 3,2%‐7,6%).
DiscussãoNo presente estudo foram incluídas 461 crianças com hemoglobinopatia SC. A gravidade das manifestaço¿es cli¿nicas na doença SC foi variável de criança para criança. Como demonstram os limites máximos de incidência dos diversos eventos clínicos, várias crianças apresentaram valores elevados, sugeriram gravidade de curso clínico semelhante à que se observa em crianças SS.
Aproximadamente 75% dos pacientes apresentaram pelo menos uma CVO, resultado semelhante aos 78% encontrados por Tuttle & Koch,11 mas superior ao relatado por Powars et al.,12 com 60% dos casos. A incidência de CVO para toda a população avaliada no presente estudo foi de 51 crises por 100 pacientes‐ano. A coorte americana apresentou incidência média de 30 CVO por 100 pacientes‐ano.13 O mesmo grupo observou que o tempo para o primeiro relato de dor na infância foi influenciado pelo genótipo da doença, foi significativamente mais precoce na doença SS (mediana de 13,9 meses) do que na doença SC (mediana de 43,6 meses).14 No presente estudo, a mediana foi de 61 meses. Crises dolorosas nas crianças com hemoglobinopatia SC ocorrem com cerca da metade da freque¿ncia do que se observa na anemia falciforme.2,5,12,13 Contudo, cabe ressaltar que apesar de a incidência ser mais baixa, ela é relevante e deve ser destacada para as equipes de saúde em unidades de urgência, com o objetivo de se evitar o subtratamento e a desvalorização das queixas de dor em pacientes com hemoglobinopatia SC.
Sobre a avaliação de órgãos‐alvo, foi verificada anotação compatível com retinopatia em 20,3% crianças que fizeram fundoscopia. A maior parte dos estudos sobre retinopatia foi feita em pacientes adultos, com prevalência média de aproximadamente 33%.2,3,15 Entre os estudos que incluem crianças, o resultado foi semelhante ao encontrado por dois grupos americanos com 23% e 16,4%, respectivamente.13,16 Sobre a avaliação cardiológica, 4,6% das crianças que fizeram ecocardiograma apresentaram valores ecocardiográficos sugestivos de hipertensão pulmonar, resultado semelhante ao encontrado por estudo inglês,17 com 6%. Contudo, o que chama a atenção no presente estudo foi o pequeno número de crianças que fizeram fundoscopia (25%) e ecocardiograma (28,2%), uma vez que os pacientes deveriam ser encaminhados para avaliações oftalmológicas e cardiológicas preventivas anuais após os cinco anos. É possível que as incidências reais de retinopatia falciforme e de alterações ecocardiográficas sugestivas de hipertensão pulmonar na população investigada estejam subestimadas. Isso demonstra a necessidade de se estabelecer rede permanente de interconsultores experientes em retinopatia proliferativa e cardiologia infantil para detecção e tratamento precoces adequados.
Quanto à avaliação ortopédica, foi encontrada descrição de necrose avascular em 58,3% dos pacientes avaliados. Na literatura, a prevalência varia entre 8,8 e 15%,3,12,13,18 mas o pequeno número de casos da presente casuística prejudica conclusão sólida. Quatro meninos (0,9%) tiveram episódios de priapismo durante o período de acompanhamento. Os dados publicados sobre priapismo geralmente resultam da análise conjunta dos genótipos SS, SC e S‐beta talassemia, com prevalência aproximada de 35% em adultos.19 Da mesma forma, o pequeno número de casos e o desenho retrospectivo do estudo limitam a análise dos dados.
Sessenta e oito crianças (14,8%) apresentaram SEA. Existem poucos estudos sobre SEA que envolveram crianças com hemoglobinopatia SC. A prevalência situa‐se entre 5 e 12%.4,20,21 A maior parte dos primeiros episódios de SEA do presente estudo (70%) ocorreu até cinco anos de vida. Estudo feito pelos mesmos autores em 255 crianças SS/Sß0 talassemia atendidas no Hemocentro de Belo Horizonte mostrou que, além de a incidência de SEA ser aproximadamente cinco vezes mais elevada nas crianças SS/Sß0 talassemia (10,2 primeiros episódios por 100 pacientes‐ano), 75% delas apresentaram o primeiro evento antes dos dois anos.22 Houve convergência na indicação de esplenectomia após dois ou mais episódios de SEA em ambos os estudos. A maioria dos autores, entretanto, recomenda o procedimento cirúrgico após o primeiro ou, no máximo, após o segundo episódio de SEA.18,21 A nosso ver, pelos dados apresentados, os cuidados com a educação dos cuidadores de criança com doença SC em relação à palpação do baço se aplicam, portanto, da mesma forma que nas crianças com doença SS.
Em relação aos exames laboratoriais hematológicos basais (tabela 3), as médias de hemoglobina total, leucometria, contagem de reticulócitos e plaquetometria foram 10,5g/dL, 11.236/mm3, 3,4% e 337.098/mm3, respectivamente. A anemia na hemoglobinopatia SC é geralmente mais leve do que em pacientes SS, com valores médios de hemoglobina que variam entre 10,5 e 11,5g/dL.3,23 Assim como previamente descrito, os reticulo¿citos esta¿o ligeiramente aumentados e a contagem de plaquetas e de leuco¿citos, normais.2,5
Todas as crianças que fizeram DTC apresentaram baixo risco de acidente vascular cerebral isquêmico (tabela 1). Sabe‐se que a incidência de acidente vascular cerebral isquêmico é aproximadamente quatro vezes menor do que nos pacientes com hemoglobinopatia SS, com taxas entre 0,8 e 3%.12,24 Atualmente não existem estudos e dados suficientes a respeito dos valores de referência da VMMmax para os pacientes com hemoglobinopatia SC, é usada a mesma classificação dos pacientes com hemoglobinopatia SS.18,24,25 Vieira et al.26 relataram 0,7% de exames com alto risco de AVC em 558 crianças com hemoglobinopatia SC. Esses autores sugerem que deveriam ser usados valores de referência mais baixos nas crianças com SC para que sejam caracterizadas como de alto risco para AVCi, mas isso demanda estudos adicionais.
Uma criança do presente estudo apresentou episódio de AVCi. Deane et al. descreveram um paciente com hemoglobinopatia SC que teve AVCi aos cinco anos, considerado secundário à doença de base após descartadas outras possibilidades; a VMMax era de 146cm/s na artéria cerebral média direita e 126cm/s na artéria cerebral média esquerda.27 Um caso de AVC em criança de sete anos foi também descrito por Powars et al.,28 em 1978, e outro, em criança nigeriana de seis anos, por Lagunju e Brown,29 em 2012. Três casos pediátricos foram assinalados por Ohene‐Frempong no estudo cooperativo americano.24 Como já referido, não existem dados atuais sobre valores de VMMax para que seja feita a estratificação de risco de AVCi em pacientes com hemoglobinopatia SC. Portanto, os resultados de VMMax do presente estudo (tabela 1) contribuem para preencher algumas lacunas no entendimento da vasculopatia cerebral das crianças SC.
O uso de hidroxiureia causou significativa redução dos sintomas, semelhante à descrito previamente.7–9 Nota‐se que nenhum paciente atingiu a dose máxima de hidroxiureia (35mg/Kg/dia), devido à superveniência de toxicidade hematológica. Luchtman‐Jones et al. relataram 133 crianças com hemoglobinopatia SC tratados com hidroxiureia, em 18 centros de referência americanos. O estudo mostrou redução de 38% dos eventos dolorosos após 12 meses de uso da medicação.9
A probabilidade estimada de óbito aos 10 anos, por todas as causas, foi de 5,4%, bem abaixo de 13,7% registrada para crianças SS/Sß0 talassemia e semelhante aos 4,4% registrados em estudo que abarcou todo o Estado de Minas Gerais.30 O grupo cooperativo americano relatou taxa de mortalidade de 9% entre 284 pacientes da coorte.13 Observa‐se que a maior parte das crianças (65,2%) faleceu com idade inferior a cinco anos. Essa constatação pode ser explicada pela maior incidência, na faixa etária descrita, de eventos agudos graves e de alto potencial de letalidade, tais como infecções e SEA, causas mais frequentes de óbito no presente estudo. Nota‐se, ainda, a presença significativa de óbitos secundários a causas indeterminadas (26%), o que corrobora haver dificuldade das equipes de saúde de identificar as possíveis intercorrências clínicas secundárias à doença falciforme.
As principais limitações do presente estudo incluem a análise retrospectiva e o pequeno número de dados sobre consultas especializadas dos pacientes envolvidos, o que restringe as conclusões sobre a incidência de algumas complicações clínicas, como a retinopatia proliferativa e a necrose da cabeça de ossos longos. A avaliação sobre os efeitos da hidroxiureia também sofre limitação de amostragem. A principal razão da perda amostral de 19% (108/569) no seguimento clínico refere‐se à dificuldade para coleta do TCLE, representada por 72% das perdas. Análise de dados epidemiológicos como sexo, idade e número de consultas registradas nos prontuários do hemocentro não indica que essas crianças teriam curso clínico diferente das 461 crianças incluídas no estudo, mas essa possibilidade não pode ser descartada. Embora tenha sido incluídas apenas crianças de Minas Gerais, é possível que os resultados apresentados sejam extrapoláveis para outros estados, pois o alelo C tem origem unicêntrica,2 embora outros fatores genéticos e ambientais possam atuar de forma diversa em crianças de outros estados e países.
Em conclusão, a análise dos resultados acrescenta informações relevantes sobre o perfil do comportamento clínico e laboratorial da doença da hemoglobina SC em nosso meio, com vistas a ações de saúde preventivas e curativas. Complicações agudas e lesões de órgãos‐alvo ocorrem em graus diversos e várias crianças apresentam manifestações clínicas intensas, semelhantes às da hemoglobinopatia SS.
FinanciamentoFundação Hemominas, Newborn Screening Program (Nupad‐UFMG), Fundação de Amparo à Pesquisa de Minas Gerais (Fapemig) e Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Ao CNPq, Fapemig e Nupad pelo apoio financeiro ao projeto. Aos colegas da Fundação Hemominas pelo apoio logístico. Às crianças e parentes pelo assentimento para feitura da pesquisa e publicação dos resultados.
Como citar este artigo: Rezende PV, Santos MV, Campos GF, Vieira LL, Souza MB, Belisário AR, et al. Clinical and hematological profile in a newborn cohort with hemoglobin SC. J Pediatr (Rio J). 2018;94:666–72.




