


To show the general prevalence and to characterize tetrahydrobiopterin (BH4) deficiencies with hyperphenylalaninemia, identified by the Neonatal Screening Program of the State of Minas Gerais.
MethodsDescriptive study of patients with BH4 deficiency identified by the Neonatal Screening Program of the State of Minas Gerais.
ResultsThe prevalence found was 2.1 for 1,000,000 live births, with a frequency of 1.71% among hyperphenylalaninemias. There were four cases (40%) with 6-pyruvoyl-tetrahydropterin synthase deficiency, three with GTP cyclohydrolase I – autosomal recessive form deficiency, and three with dihydropteridine reductase deficiency (30% each). Six patients were diagnosed due to clinical suspicion and four cases due to systematic screening in neonatal screening. After the start of the treatment, patients identified by neonatal screening had rapid improvement and improved neuropsychomotor development compared to those diagnosed by the medical history.
ConclusionsThe prevalence of BH4 deficiencies in Minas Gerais was slightly higher than that found in the literature, but the frequency among hyperphenylalaninemias was similar. Although rare, they are severe diseases and, if left untreated, lead to developmental delays, abnormal movements, seizures, and premature death. Early treatment onset (starting before 5 months of age) showed good results in preventing intellectual disability, justifying the screening of these deficiencies in newborns with hyperphenylalaninemia identified at the neonatal screening programs for phenylketonuria.
Apresentar a prevalência geral e caracterizar as deficiências de tetrahidrobiopterina - BH4 - com hiperfenilalaninemia, identificadas pelo Programa de Triagem Neonatal do Estado de Minas Gerais.
MétodosEstudo descritivo de pacientes com deficiência de BH4 do Programa de Triagem Neonatal do Estado de Minas Gerais.
ResultadosA prevalência encontrada foi de 2,1 para 1.000.000 recém-nascidos vivos e a frequência de 1,71%, dentre as hiperfenilalaninemias. Quatro casos (40%) com deficiência de 6-piruvoil-tetrahidropterina sintase, três com deficiência de GTP ciclohidrolase I e três com deficiência de dihidropteridina redutase (30% cada um). Seis pacientes foram diagnosticados por suspeita clínica e quatro pela pesquisa sistemática na triagem neonatal. Após o início do tratamento, os pacientes identificados pela triagem neonatal tiveram melhora rápida e melhor desenvolvimento neuropsicomotor em comparação com aqueles diagnosticados pela história clínica.
ConclusõesA prevalência das deficiências de BH4 em Minas Gerais foi um pouco maior que a encontrada na literatura, mas a frequência, entre as hiperfenilalaninemias, foi semelhante. Embora raras, são graves e, se não tratadas, levam a atraso de desenvolvimento, movimentos anormais, convulsões e morte precoce. O tratamento precoce (início antes dos 5 meses) mostrou bons resultados na prevenção de deficiência intelectual, justificando a pesquisa dessas deficiências nos recém-nascidos com hiperfenilalaninemia pelos programas de triagem neonatal para fenilcetonúria.
Hyperphenylalaninemias are manifestations of the genetic defects most frequently involved in amino acid metabolism.1 To date, five defects are known to lead to hyperphenylalaninemia states: defect in the phenylalanine hydroxylase enzyme, causing phenylketonuria, and in four different enzymes involved in the synthesis or regeneration of tetrahydrobiopterin (BH4), which is an important cofactor of the enzymes involved in the synthesis of tyrosine, dopamine, serotonin, nitric oxide, and glycerol.2
The four BH4 deficiencies that occur with hyperphenylalaninemia are deficiency of GTP cyclohydrolase I – autosomal recessive form (GTPCH I), 6-Pyruvoyl-tetrahydropterin synthase (PTPS) deficiency, dihydropteridine reductase (DHPR) deficiency, and pterin-4α-carbinolamine dehydratase (PCD) deficiency. They account for approximately 2% of cases of hyperphenylalaninemia, with a worldwide prevalence of 1 per 1,000,000 live births.3
As BH4 is involved in the synthesis of neurotransmitters such as dopamine and serotonin, deficiencies of this cofactor can lead not only to hyperphenylalaninemia states but also neurological symptoms and signs, resulting from the deficiency of these neurotransmitters.4
Since these deficiencies occur with hyperphenylalaninemia, their identification is possible through the neonatal screening programs for phenylketonuria, allowing the diagnosis to be made in the first weeks of life5,6 and prompt treatment to be initiated. The current routine of investigating BH4 deficiencies in every newborn with hyperphenylalaninemia by analyzing pterins and DHPR enzyme activity in blood samples on filter paper, then starting early treatment, has resulted in better intellectual levels in affected patients than were obtained previously to this routine.7
The treatment of BH4 deficiencies should be individualized due to the great inter-individual allelic and non-allelic heterogeneity, a characteristic of the disease.4
In Brazil, neonatal screening for hyperphenylalaninemias started in the 1980s. In 2001, the National Neonatal Screening Program was created and since then, neonatal screening for hyperphenylalaninemias has been expanded and now includes the entire country.8 However, most of the states do not systematically screen for BH4 deficiencies, which leads to delayed diagnosis and treatment onset.
In the state of Minas Gerais, the Neonatal Screening Program (NSPMG) is coordinated by the Action and Research Center in Diagnostic Support (Núcleo de Ações e Pesquisa em Apoio Diagnóstico [NUPAD]) and includes the 853 municipalities. Since its implementation, it has already screened more than 4,300,000 newborns. It aims at the early diagnosis and treatment of sickle cell anemia, cystic fibrosis, congenital hypothyroidism, biotinidase deficiency, and phenylketonuria. Since 2006, it has systematically carried out the screening for BH4 deficiency. It is, to the best of the authors’ knowledge, the location with the largest number of patients in longitudinal follow-up with these deficiencies in Brazil.
Due to their rarity, BH4 deficiencies are still poorly understood and there are few studies involving patients with this diagnosis.7,9–11
Some signs and symptoms have been described, but the phenotype as a whole is still little known, as well as its clinical evolution. As in Brazil most of the neonatal screening programs for phenylketonuria do not screen for BH4 deficiencies, the knowledge of the clinical symptoms and the evolution of these patients may contribute to earlier diagnostic suspicion and to anticipate diagnostic and therapeutic measures, improving quality of life. For close relatives, genetic counseling is of great importance in family planning.
The present study aimed to identify the types, general prevalence, and frequency of these deficiencies in newborns with hyperphenylalaninemias screened by NSPMG from its implementation in 1993 until December 2012. Its proposal was to describe the several clinical characteristics of the disease, allowing a better understanding.
MethodsA cross-sectional and retrospective study was carried out, after its approval by the Ethics Committee of Universidade Federal de Minas Gerais (UFMG) (CAAE – 04096512.0.0000.5149), which identified all patients screened by the NSPMG, diagnosed with one of the forms of BH4 deficiency that develops with hyperphenylalaninemia. These diagnoses were performed by the Biochemistry Laboratories of the University Children's Hospital Zürich, in Switzerland, and the Faculté Libre de Medicina, in France, based on the analysis of pterins and the DHPR activity in blood and/or urine sample collected on the day of the first consultation of patients in the Phenylketonuria Outpatient Clinic of Hospital das Clínicas of UFMG and deposited on filter paper that, after dried, was sent by mail.
The calculation of the general prevalence of BH4 deficiencies was obtained from the ratio between the number of diagnosed cases, since the implantation of the Screening Program until December 2012, and the number of newborns screened in the same period. When calculating the frequency of BH4 deficiencies among the hyperphenylalaninemias, the denominator used was the number of cases of hyperphenylalaninemias identified in the same period.
After identifying the patients with the deficiencies, their clinical and laboratory characteristics up to December 2012 were recorded, such as delayed neuropsychomotor development, hypotonia, myoclonus, spasticity, seizures, hyperreflexia, hypersalivation, serous coryza, fine tremor, irritability, and serum phenylalanine levels. The time elapsed between treatment onset and symptom improvement or disappearance was an important factor in the comparison between the patients who had been diagnosed by clinical suspicion and those diagnosed by the systematic investigation of BH4 deficiencies.
Data collectionThe databases of the Action and Research Center in Diagnostic Support of Faculdade de Medicina da Universidade Federal de Minas Gerais (NUPAD/FM/UFMG), a referral service of the Screening Program in the State of Minas Gerais, and of the Phenylketonuria Outpatient Clinic of the Special Genetics Service of Hospital das Clínicas of UFMG, were accessed, where data from all patients screened and those who are followed are stored. Patients’ charts were also consulted. The parents of patients with BH4 deficiency signed the informed consent authorizing study participation.
Inclusion criteriaAll patients with hyperphenylalaninemia due to BH4 deficiency, confirmed by pterin analysis and DHPR activity, were included in the study.
ResultsFrom September 1993 to December 2012, 4,684,515 newborns were screened in Minas Gerais, Brazil. Of these, 586 were identified with hyperphenylalaninemia, of whom 10 were due to BH4 deficiency, eight of whom were males (80%).
The prevalence found was 2.1 for every 1,000,000 live births and the frequency was 1.71% among the hyperphenylalaninemia cases.
All patients were followed through regular consultations, whose frequency varied from two to six months.
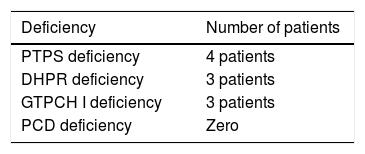
The distribution, by type of deficiency, is shown in Table 1.
Three patients died before 6 years of age: two with PTPS deficiency (at 5 and at 10 months of age) and one with GTPCH I deficiency (at 5 years of age). These were deaths without known cause and occurred in their homes.
The first six patients, all before 2006, were initially diagnosed as phenylketonuria cases, but did not adequately respond to dietary treatment, and started to show clinical signs suggestive of BH4 deficiencies. After 2006, when the systematic screening for BH4 deficiencies was established, four more patients were identified.
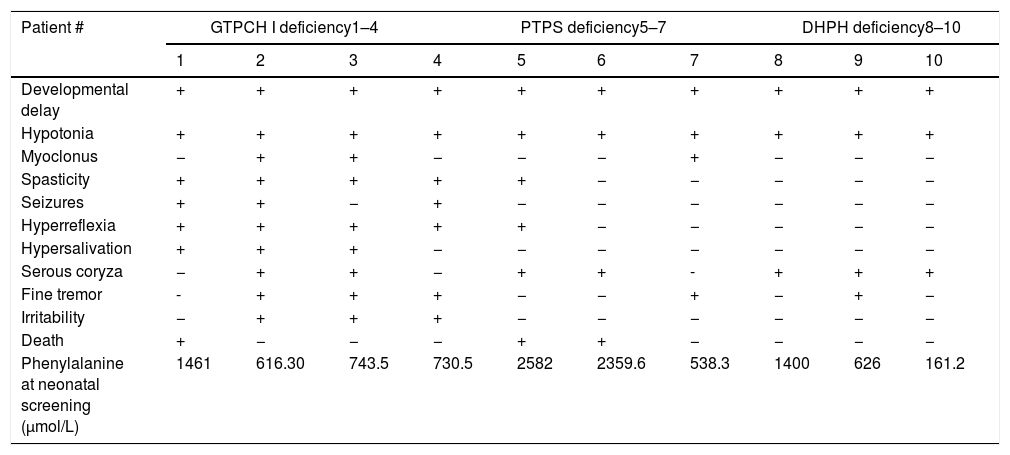
Table 2 summarizes the signs and symptoms of each patient, according to the type of deficiency. These clinical characteristics emerged before treatment started and none after treatment was implemented.
Signs and symptoms found in patients with BH4 deficiency in Minas Gerais: (+) present symptoms (−) absent symptoms.
| Patient # | GTPCH I deficiency1–4 | PTPS deficiency5–7 | DHPH deficiency8–10 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Developmental delay | + | + | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + | + | + | + | + | + | + | + |
| Myoclonus | − | + | + | − | − | − | + | − | − | − |
| Spasticity | + | + | + | + | + | − | − | − | − | − |
| Seizures | + | + | − | + | − | − | − | − | − | − |
| Hyperreflexia | + | + | + | + | + | − | − | − | − | − |
| Hypersalivation | + | + | + | − | − | − | − | − | − | − |
| Serous coryza | − | + | + | − | + | + | - | + | + | + |
| Fine tremor | - | + | + | + | − | − | + | − | + | − |
| Irritability | − | + | + | + | − | − | − | − | − | − |
| Death | + | − | − | − | + | + | − | − | − | − |
| Phenylalanine at neonatal screening (μmol/L) | 1461 | 616.30 | 743.5 | 730.5 | 2582 | 2359.6 | 538.3 | 1400 | 626 | 161.2 |
Tables 3 and 4 show (for patients with a diagnosis based on clinical suspicion and for patients diagnosed by neonatal screening, respectively) the ages of the first symptoms, at the diagnosis and treatment onset, and at the beginning of clinical improvement.
Age at onset of symptoms/signs, diagnostic confirmation, and clinical improvement onset in patients with BH4 deficiency, identified based on the clinical suspicion of the disease.
| BH4 deficiency (patient #) | Symptom onset | Diagnostic confirmation and age at start of the treatment | Onset of clinical improvement (months after start of the treatment) |
|---|---|---|---|
| GTPCH I#1 | 2 months | 11 months | 4 months (died at 5 years of age) |
| GTPCH I#2 | 4 months | 9 months | 2–3 months |
| GTPCH I#3 | 2 months (severe symptoms) | 5 months | 2 months |
| PTPS#4 | 3 months | 8 years | 5 months |
| PTPS#5 | 4 months | Died at 5 months of age | |
| PTPS#6 | 2 months | 5 months | 2 months (died at 10 months of age) |
Age at onset of symptoms/signs, diagnostic confirmation, and clinical improvement onset in patients with BH4 deficiency, identified at the neonatal screening for the disease.
| BH4 deficiency (patient #) | Symptom onset | Diagnostic confirmation and age at the start of the treatment | Clinical improvement onset (months after the start of the treatment) |
|---|---|---|---|
| PTPS #7 | 3 months | 4 months | 1 month |
| DHPR #8 | 4 months | 5 months | 1 month |
| DHPR #9 | 2 months | 2 months | 1 month |
| DHPR #10 | 2 months | 4 months | 1 month |
The frequency of BH4 deficiencies varies greatly worldwide. More than half of patients are Caucasians (52.9%); 37.2% are Turks; 12% are Arabs; 10.4% are Chinese; 2.1% are Indians; 2.7% are Japanese.3 However, it is a very rare disease, corresponding in most countries to 2% of screened hyperphenylalaninemias.1
The phenotype description of the several types of BH4 deficiency is important, since it is a rare disease with a prevalence of 1:106 and with few descriptions of patients, especially during long observation periods.7,9–11 In Brazil, this is even more important, since pterin analyses are not yet performed in the country and the diagnosis depends on sending the biological material abroad. This fact makes several states that carry out the neonatal screening for phenylketonuria depend on clinical evaluation for diagnosis and treatment.
In a literature review, only two case reports described in Brazil were found, both in the state of Rio Grande do Sul, by the same team. In 1983, Giugliani et al. described two patients with hyperphenylalaninemia who did not progress well with the dietary treatment for phenylketonuria. After the pterin analysis, the diagnosis of BH4 deficiency was confirmed.12 In 1994, Jardim et al. described five cases of PTPS deficiency in patients with a diagnostic suspicion of inborn errors of metabolism.13
The present study was carried out at the Phenylketonuria Outpatient Clinic of the Special Genetics Service of Hospital das Clínicas of Universidade Federal de Minas Gerais, where all patients with hyperphenylalaninemia in the state of Minas Gerais are followed.
Until 2006, the identification of BH4 deficiencies was performed based on clinical suspicion in patients who did not have a satisfactory response to dietary treatment for phenylketonuria, despite adhering to it. From that year on, screening for BH4 deficiencies started to be systematically performed, with pterin measurement and assessment of DHPR enzyme activity in all patients screened with hyperphenylalaninemia, in the first consultation at the service. It is possible that some were lost because they had phenylalanine levels below the cutoff value used (240μmol/L or 4mg/dL). However, as patients with transient hyperphenylalaninemia are followed for six months and those with permanent hyperphenylalaninemia up to six years, with girls returning at puberty,14 the losses are likely to be minimal.
After 2006, with the systematic screening for BH4 deficiencies in all patients with hyperphenylalaninemia, the losses, if any, should have been even lower. Possible biases, especially regarding research, may have occurred due to the small number of cases found. Nonetheless, considering the total number of screened patients and the lack of sample selection, it is possible that the results are representative of the population.
The prevalence found was slightly higher than that described; however, the percentage of patients among the cases of hyperphenylalaninemia was similar to that described in the literature.1,15,16 In 1994, Jardim et al.,13 in the state of Rio Grande do Sul, Brazil, studying patients with suspected inborn errors of metabolism, found a very high prevalence of PTPS deficiency. This is explained by the fact that it is a study of selected patients with suspected diagnosis of related diseases. Although the present study population was large (4,684,515 screened patients) and the prevalence (2.1:106) higher than that described, the number of cases was relatively small. Nevertheless, to the best of the authors’ knowledge, it represents the largest number of cases with longitudinal follow-up in Brazil.
Similar to what was described in the literature,3 PTPS deficiency was the most common in the present study, although with a lower percentage (40%) than described (approximately 60%).3 After that, the most frequently identified deficiencies were DHPR (30%) and GTPCH I (30%). Although the first deficiency showed a frequency similar to the one described,3,17 the second showed a much lower percentage (4–5%) in the literature.3,17 No cases of PCD deficiency were identified, which the literature reports as rare (3–4% of cases).3,17
Symptom onset in all patients with BH4 deficiency occurred between two and four months, as described in the literature.1,3,17–19 No difference was observed regarding the age at symptom onset of the different types.
In eight patients, phenylalanine levels at the screening were compatible with the diagnoses of mild or classical phenylketonuria. Patient # 7 had a phenylalanine level compatible with the probable diagnoses of transient or permanent hyperphenylalaninemia. Patient #10, however, had a phenylalanine level below the cutoff value and would not have been diagnosed if her twin (patient # 9) had not been, too. These facts warn that BH4 deficiencies may show low phenylalanine levels at the neonatal screening and reinforce the need for its investigation in all patients, even those with slightly increased phenylalanine levels.20,21 They also alert to the possibility of false negative results, even when the screening is used for diagnosis.
All signs and symptoms appeared before treatment was implemented. All patients showed developmental delay and muscle hypotonia, as described in the literature.22 The third most frequent sign was serous coryza (70% of patients), followed by fine tremor, hyperreflexia, and spasticity, present in 50% of the patients. Irritability, myoclonus, seizures, and hypersalivation were present in 30% of the patients. These data, all described in the literature,19,23–28 warn that the combination of signs and symptoms may be variable, and that neurological assessment helps in the diagnostic suspicion.
Death occurred in 30% of patients, a higher rate than described (approximately 13%),3 demonstrating the disease severity.18,29 Of these patients, two had PTPS deficiency and one had GTPCH I deficiency. One patient died before it was possible to start the therapy. The exact cause of death is unknown. In the literature, cases of death due to BH4 deficiency are attributed to sudden death, with no precise specification of the cause.3
All patients who died were diagnosed before the systematic study of the pterins and DHPR activity.
Patients who were diagnosed after clinical suspicion had confirmation and started treatment later, between 5 and 11 months. Despite this, treatment showed good results, especially in relation to the dystonic pictures, even in a patient with PTPS deficiency (#4), whose diagnosis was made eight years after symptom onset. He had a severe neurological condition, but currently, he can walk, speak with difficulty, and maintain good interaction with the environment, although he is still very dependent regarding activities of daily living.
This patient had a suspected diagnosis since he was five months old; however, his mother did not want to continue the investigation; she had another child, equally affected. She only returned to the clinic eight years later, due to the eldest son's death. This fact alerts to the importance of the family's emotional reaction when an early diagnosis is not attained and the patient does not respond well to the implemented therapy.
Treatment response was somewhat slower in patients whose diagnosis was made after clinical suspicion. These patients, despite a good clinical response, persisted with some degree of developmental delay, demonstrating early brain impairment. It is suspected that neuronal development, early functional synapse maturation, and myelination are altered by BH4 deficiencies during critical periods of brain development.18
Patients whose diagnoses were obtained after the systematic study of pterins and DHPR activity had a shorter time between treatment onset and the start of symptom improvement (approximately one month). They also showed better neuropsychomotor development, and two of them (#7 and #8) had an apparently normal development until the end of this study. Two female twin patients (#9 and #10), despite good motor development, have been showing speech delay. Their mother's pregnancy was complicated by preeclampsia, toxoplasmosis, and preterm labor. Due to maternal toxoplasmosis, the patients were treated for this disease up to one year of age. It is important to consider the overlap of toxoplasmosis with speech difficulty.
The early treatment of BH4 deficiencies is only possible if the differential diagnosis of the hyperphenylalaninemia types is performed as soon as possible, preferably within the first month of life.11 Although treatment is yet to be started within this period, since the diagnosis is still made outside of the country, the satisfactory evolution of the diagnosed patients justifies this recommendation.
Patients with DHPR deficiency showed the best clinical outcome, although this is described as the most severe form of BH4 deficiency.22 Early treatment probably contributed to this finding.
No patient showed any adverse drug reactions.
Sapropterin (synthetic BH4) was not regularly used due to the difficulty in obtaining the medication in Brazil, since it has not been licensed by the Brazilian National Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]). Only two patients (#2 and #3) with GTPCH I deficiency used it, for a short time. The medication is available in only a few countries and its importation is very expensive. The medication acquisition was only possible through a lawsuit. In the absence of medication, hyperphenylalaninemia was controlled with a specific diet, with good results. Genetic counseling was offered to all parents.
The number of cases identified was larger than expected, but still small for more definitive conclusions. The sample size may explain the disagreements with the percentages described in the literature. This study reinforces the opinion of the European Phenylketonuria Group of March 2011, which states that after neonatal screening for phenylketonuria, every newborn, even those with slightly elevated serum phenylalanine levels, should be screened for BH4 deficiency.20
This is the group's current recommendation to neonatal screening services, supported by Han et al., who emphasize that early diagnosis and treatment influence the disease prognosis and help maximize the potential for development.30 It also emphasizes the need for these tests to be performed in Brazil, which will allow even earlier diagnosis and genetic counseling.
As these are very rare diseases, many health professionals are unaware of their characteristics and their forms of treatment. It is expected that this study will contribute to a better understanding of the clinical and epidemiological aspects of BH4 deficiencies in the state of Minas Gerais.
The results were consistent with the scientific literature. Specific mutations have already been described in a small number of cases, but genotype-phenotype associations are still not consistent. This may be an interesting line for future studies in Brazil as well.
The authors expect, in the near future, to be able to conduct the tests that identify BH4 deficiencies in Brazil, allowing even earlier diagnoses and treatments, with important improvement in the clinical evolution of affected patients.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Souza CA, Alves MR, Soares RD, Kanufre V, Rodrigues VM, Norton RC, et al. BH4 deficiency identified in a neonatal screening program for hyperphenylalaninemia. J Pediatr (Rio J). 2018;94:170–176.
Study carried out at Universidade Federal de Minas Gerais (UFMG), Hospital das Clínicas, Núcleo de Ações e Pesquisa em Apoio Diagnóstico (NUPAD), Belo Horizonte, MG, Brazil.





