





This review article aimed to present a clinical approach, emphasizing the diagnostic investigation, to children and adolescents who present in the emergency room with acute-onset muscle weakness.
SourcesA systematic search was performed in PubMed database during April and May 2017, using the following search terms in various combinations: “acute,” “weakness,” “motor deficit,” “flaccid paralysis,” “child,” “pediatric,” and “emergency”. The articles chosen for this review were published over the past ten years, from 1997 through 2017. This study assessed the pediatric age range, from 0 to 18 years.
Summary of the dataAcute motor deficit is a fairly common presentation in the pediatric emergency room. Patients may be categorized as having localized or diffuse motor impairment, and a precise description of clinical features is essential in order to allow a complete differential diagnosis. The two most common causes of acute flaccid paralysis in the pediatric emergency room are Guillain–Barré syndrome and transverse myelitis; notwithstanding, other etiologies should be considered, such as acute disseminated encephalomyelitis, infectious myelitis, myasthenia gravis, stroke, alternating hemiplegia of childhood, periodic paralyses, brainstem encephalitis, and functional muscle weakness. Algorithms for acute localized or diffuse weakness investigation in the emergency setting are also presented.
ConclusionsThe clinical skills to obtain a complete history and to perform a detailed physical examination are emphasized. An organized, logical, and stepwise diagnostic and therapeutic management is essential to eventually restore patient's well-being and full health.
O objetivo deste artigo de revisão é apresentar uma abordagem clínica, enfatizando a investigação diagnóstica, voltada para crianças e adolescentes no pronto socorro com fraqueza muscular de surgimento agudo.
FontesFoi realizada uma pesquisa sistemática na base de dados PubMed entre abril e maio de 2017, utilizando os seguintes termos de pesquisa em várias combinações: “agudo”, “fraqueza”, “déficit motor”, “paralisia flácida”, “criança”, “pediátrico” e “emergência”. Os trabalhos escolhidos para esta revisão foram publicados nos últimos dez anos, de 1997 a 2017. Este trabalho aborda a faixa etária pediátrica, de 0 a 18 anos.
Resumo dos dadosO déficit motor agudo é uma causa razoavelmente comum para crianças e adolescentes procurarem o pronto socorro. Os pacientes podem ser classificados como apresentando deficiência motora localizada ou difusa, e uma descrição precisa das características clínicas é essencial para possibilitar um diagnóstico diferenciado completo. As duas causas mais comuns de paralisia flácida aguda no pronto socorro pediátrico são síndrome de Guillain-Barré e mielite transversa, independentemente de outras etiologias serem consideradas, como encefalomielite disseminada aguda, mielite infecciosa, miastenia grave, derrame, hemiplegia alternante da infância, paralisia periódica, encefalite do tronco encefálico e fraqueza muscular funcional. Os algoritmos da investigação de fraqueza aguda localizada ou difusa na configuração de emergência também são apresentados.
ConclusõesSão enfatizadas as habilidades clínicas para obter um histórico completo e realizar um exame físico detalhado. Um manejo diagnóstico e terapêutico organizado, lógico e por etapas é essencial para eventualmente restaurar o bem-estar e a saúde total do paciente.
Acute motor deficit or weakness is a fairly common presentation in the pediatric emergency room (ER). Across all age ranges, 5% of all patients visiting the ER have neurologic symptoms.1 In this setting, the pediatrician must be comfortable in performing the initial workup, as some etiologies may be life-threatening and demand urgent care. Timely interventions are likely to restore patient's well-being and full health.
Weakness is categorized as a negative motor sign, as well as ataxia and apraxia. It is valuable to have a firm grasp on the definition of these three negative motor signs. Weakness is defined as the inability to generate normal voluntary force in a muscle or normal voluntary torque in a joint, while ataxia is the inability to generate a normal voluntary movement trajectory that cannot be attributed to weakness or involuntary muscle activity in the affected joints.2 Ataxic movements are uncoordinated and clumsy, but there is no underlying weakness of the involved muscles. Apraxia is the inability to perform previously learned complex movements, which is not explained by weakness, ataxia, or involuntary motor activity, i.e., the motor, sensory, basal ganglia, and cerebellar functions are intact.2 It is noteworthy that sensory weakness does not exist.
For the purpose of this study, plegia will be used to denote complete or partial weakness,3 but the reader is informed that strictly speaking plegia means complete paralysis and paresis implies that muscle strength is only partially affected. A common descriptor of acute weakness is acute flaccid paralysis, meaning that in the ER setting, paralysis is usually not accompanied by spasticity or other abnormal signs of central nervous system (CNS) motor tracts, e.g., hyperreflexia, clonus, or Babinski reflex.4
This study aimed to present a clinical diagnostic approach to children and adolescents who present with an acute motor deficit in the ER and to review the most frequent etiologies.
MethodsA systematic search was performed in PubMed database during April and May 2017, using the following search terms in various combinations: “acute,” “weakness,” “motor deficit,” “flaccid paralysis,” “child,” “pediatric,” and “emergency”. The articles chosen for this review were published over the past ten years, from 1997 through 2017. This study assessed the pediatric age range, from 0 to 18 years.
Clinical features and differential diagnosisAcute weakness may present either as a localized or diffuse impairment. A precise description of the motor deficit — its mode of onset, duration, and progression — is essential to allow a complete differential diagnosis. In this sense, a detailed history and a full physical examination, including an objective neurologic examination, provide the best opportunities to expedite etiology identification. Table 1 outlines the essential information to be compiled during the initial approach of a patient presenting with acute weakness.
History and physical examination of a patient with acute motor deficit.
| History |
| Is this the first time the child presents with weakness? |
| Onset – where was the child and what was he/she doing when the deficit was detected? |
| Rhythm – abrupt, rapidly or slowly progressing, fluctuating or remitting–recurring? |
| Is the weakness associated with any precipitating factors, such as a place, a person, a food, a medication, or a time of the day? |
| Does the weakness improve with rest and worsen with activity (fatigability)? |
| Has there been an increased frequency of falls in the recent past? |
| Has the weakness progressed in an ascending or descending mode? |
| Associated symptoms of infection (fever, headache, vomiting, diarrhea) or of a concurrent disease |
| Preceding or concomitant sensory symptoms, such as pain/tenderness, dysesthesias, numbness, headache, or visual disturbances |
| Changes in urination and bowel habits |
| Mental status changes – has there been any impairment in patient's mental status? |
| History of recent infections and other illnesses |
| Use of all medications, including herbal medicine, eye drops, etc. |
| Developmental history – ages of motor skills acquisition, any regression, other areas of development |
| History of trauma, especially to the head and back |
| History of seizures |
| Contact with other ill individuals |
| Psychosocial history – recent stress, interactions with peers, school performance, behavioral issues, death or departure of loved ones |
| Contact with pets and other animals |
| Physical and neurologic examination |
| Vital signs |
| Head circumference |
| Growth parameters |
| Body symmetry |
| Nuchal rigidity and other meningeal signs |
| Respiratory distress signs |
| Mental status – alertness, irritability, orientation to time and place, language skills |
| Cardiovascular and respiratory systems and abdomen examination |
| Cranial nerve examination – pupils’ size and reaction to light? Visual acuity? Funduscopy? Facial weakness? Extraocular muscle movements? Palate and tongue movements? |
| Motor examination – muscle tonus and strength, deep tendon reflexes, ankle clonus, pronator-drift test |
| Babinski sign |
| Sensory examination searching for a sensory level |
| Coordination – finger-to-nose test, diadochokinesia |
| Gait – is the child able to sit or to walk without/with support? Walking on tiptoes, on ankles |
| Signs of a chronic muscle disease, e.g., muscle atrophy, fasciculations, myotonia, Gowers’ sign |
The pattern through which weakness extends to other body parts may offer clues to diagnosis. For example, a relatively symmetric, ascending deficit suggests Guillain–Barré syndrome (GBS), while a descending paralysis raises suspicion of botulism.5
Some patients will present to the ER with what appears to be acute weakness, but a thorough assessment will reveal that in fact he/she has pseudoparalysis, i.e., muscle strength is preserved and the motor function is impaired by another mechanism, most often severe pain. Common pediatrics examples of this situation are inability to walk due to calf pain secondary to acute viral myositis, pyogenic arthritis-related arthralgia (e.g., gonococcal arthritis in teenagers), nursemaid's elbow, or bone pain secondary to osteomyelitis, bone fracture, or dislocation.
Since the eradication of poliomyelitis, the two most common causes of acute flaccid paralysis in the ER are GBS and acute transverse myelitis.6 Among other etiologies (Table 2), the most relevant for the pediatric age group are acute disseminated encephalomyelitis (ADEM), infectious myelitis, myasthenia gravis (MG), stroke, alternating hemiplegia of childhood, periodic paralyses, brainstem encephalitis, and functional muscle weakness.
Etiologies of an acute diffuse motor deficit.
| Most common etiologies |
| Guillain–Barré syndrome |
| Transverse myelitis |
| Other etiologies |
| Acute disseminated encephalomyelitis (ADEM) |
| Alternating hemiplegia of childhood |
| Atlantoaxial dislocation |
| Botulism |
| Brain abscess |
| CNS infections, e.g., acute meningitis |
| CNS mass lesions |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) |
| Complicated migraine |
| Congenital myopathies |
| Epilepsy, i.e., Todd's palsy |
| Familial spastic paraplegia |
| Functional muscle weakness (formerly called conversion disorder) |
| Hypoglycemia |
| Hypophosphatemia |
| Hopkins syndrome (associated with acute asthma) |
| Infectious myelitis (e.g., by enterovirus) |
| Intracranial hemorrhage |
| Kawasaki disease |
| Metabolic disease |
| Myasthenia gravis |
| Periodic paralyses |
| Spinal cord compression |
| Vaccine-associated poliomyelitis |
| Vasculitis |
| Stroke |
CNS, central nervous system.
GBS, or acute inflammatory demyelinating polyradiculoneuropathy (AIDP), is an autoimmune disease that usually presents with acute onset of a rapidly progressive, essentially symmetric weakness and areflexia in a previously well child.7 The acute weakness in GBS tends to begin distally and to progress rostrally, and in 50–70% of cases it is preceded within four weeks by an acute respiratory or gastrointestinal infection. The etiologic agents most frequently involved are Campylobacter jejuni and Helicobacter pylori in the gastrointestinal tract, and Mycoplasma pneumoniae in the respiratory tract. An annual incidence of 0.5–2 cases per 100,000 individuals has been reported in the age range <18 years.8 In less than 10% of cases, GBS is associated with vaccination in the preceding 30 days.9
AIDP is actually a descriptive term for the most common clinical presentation of GBS, while in 10–15% of cases, the clinical features comprise variant GBS forms, such as acute motor axonal neuropathy (AMAN, more rapidly evolving and more severe motor deficits, no sensory deficits), acute motor and sensory axonal neuropathy (AMSAN, reported nearly exclusively in adults), Miller-Fisher syndrome (ataxia, ophthalmoplegia, and areflexia without peripheral weakness), pharyngeal-cervical-brachial motor variant (ptosis, facial, pharyngeal, neck flexor muscle weakness), acute pandysautonomia, and acute ophthalmoplegia.8,10,11 AMAN is due to axonal damage and is more frequently diagnosed in Asia and South America.11
Clinical onset is characterized by limb pain, limb weakness, and in a few cases ataxia. Physical examination usually shows a preserved mental status or irritability and decreased or abolished deep tendon reflexes in weak limbs.7,8 Weakness progresses in a caudo-cranial direction, but there are exceptions to this pattern. Peak weakness is usually reached after seven to ten days, but no later than four weeks. The severity of arm weakness is the most reliable factor to predict respiratory insufficiency.11
The diagnosis is based on clinical findings, and supported by albuminocytologic dissociation in cerebrospinal fluid (CSF), that is, an increased protein level despite normal leukocyte count, a spine magnetic resonance imaging (MRI) showing contrast-enhancement of nerve roots, and/or demyelination or axonal damage findings in electromyography with nerve conduction studies (EMG). Some patients have normal EMG studies in the first two weeks of illness, but later on all patients show abnormal results.6
Transverse myelitisAcute transverse myelitis (ATM) accounts for one fifth of acquired demyelinating events in children; it has an estimated annual incidence in children aged <16 years of 2 per 1,000,000, and its male:female ratio is 1.1–1.6:1.12 There are two peaks of pediatric incidence, at the age ranges 0–2 years and 5–17 years, with the highest incidence in the former.13 In two-thirds of cases, there was a prodromal infection within the past 30 days.
Affected children develop an acute onset of symmetric paraplegia or tetraplegia, decrease or loss of sensation, and sphincter dysfunction, and their clinical course extends over three phases: acute (usually two to seven days), plateau (1–26 days), and recovery phases (months to years). In some patients, pain is the presenting feature.6
ATM is a diagnosis of exclusion; therefore, it is important to carefully consider its differential diagnoses. In this regard, the diagnostic criteria established by the Transverse Myelitis Consortium Working Group may be valuable (Table 3).14
Diagnostic criteria for transverse myelitis.
| Inclusion criteria |
| Development of sensory, motor, or autonomic dysfunction attributable to the spinal cord |
| Bilateral symptoms |
| Clearly defined sensory level |
| Exclusion of compressive causes by MR imaging or CT myelography |
| Spinal cord inflammation demonstrated by cerebrospinal fluid pleocytosis, elevated IgG, or gadolinium enhancement |
| Progression to clinical nadir between 4h and 21 days from onset of symptoms |
| Exclusion criteria |
| History of irradiation to the spine within 10 years |
| Clinical findings consistent with anterior spinal artery occlusion |
| Abnormal flow voids on the surface of the cord consistent with arteriovenous malformation |
| Serologic or clinical evidence of connective tissue disease (sarcoidosis, Behçet disease, Sjögren syndrome, systemic lupus erythematosus, or mixed connective tissue disorder) |
| Central nervous system manifestations of syphilis, Lyme disease, HIV, HTLV-1, Mycoplasma, or other viral infection |
| Brain abnormalities suggestive of multiple sclerosis |
| History of clinically apparent optic neuritis |
Source: Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis.14
Children will benefit from a favorable motor outcome, with up to 56% having complete recovery; one study found that the mean time to recover the ability to walk independently was 56 days.6
Acute disseminated encephalomyelitis (ADEM)ADEM is an inflammatory, demyelinating disease that typically occurs within two days to four weeks following a viral infection or, less commonly, vaccination.15 It presents with multifocal neurologic deficits accompanied by encephalopathy, and its mean age of onset is 5.7 years, with a male:female ratio of 2.3:1.15,16
Clinical features may include fever, headache, vomiting, meningeal signs, visual loss, seizures, cranial nerve palsies, and mental status changes, ranging from lethargy to coma.15 There is an ongoing controversy on whether ADEM and multiple sclerosis (MS) belong to the same disease spectrum or are completely distinct entities.16 Although ADEM usually follows a monophasic course, its recurring and multiphasic variants blurs the distinction from MS. Brain MRI may be the most valuable tool to accomplish such distinction, as ADEM cases exhibit large areas of increased signal intensity on T2 and FLAIR-weighted images, with ill-defined borders, distributed bilaterally in the cerebral white matter (WM) and often affecting the basal ganglia, brainstem, and cerebellar and cerebral cortex gray matter, while MS cases usually show well-defined lesions that are confined to the periventricular WM.17 Absence of encephalopathy, age above 10 years, presence of optic neuritis, and intrathecal oligoclonal bands during an acute CNS demyelination event increase the risk of MS development.15
CSF may be normal or may display mild pleocytosis, with or without elevated protein levels.15
Early and aggressive treatment may attain good recovery with minimal or no deficit in over half the patients.18
Infectious myelitisAcute flaccid paralysis can result from spinal cord infection with a number of pathogens, e.g., West Nile virus, nonpolio enteroviruses, dengue virus, syphilis, Lyme disease, human immunodeficiency virus (HIV), cytomegalovirus, Epstein-Barr virus, HTLV-1, and Mycoplasma.14,19 Its main distinctive feature from transverse myelitis is that the majority of children have focal, poliomyelitis-like spinal cord paralysis with minimal or no sensory symptoms.20
Acute viral myositisAcute viral myositis manifests as muscle pain and lower-limb weakness, especially in the calves and thighs.21 It is most commonly associated with influenza virus infection. Accordingly, its prodromal symptoms include fever, headache, cough, and other respiratory signs. Affected children may refrain from moving their legs secondary to pain, or may indeed have weakness related with rhabdomyolysis.22 Among 35 cases with median age of 7.5 years, muscle weakness lasted one to eight days, and mean serum creatine-kinase was 5507U/l.21
Myasthenia gravisMG is a relatively rare autoimmune neuromuscular disorder, and its presentation in childhood is divided into transient neonatal MG and juvenile MG. The former is due to transplacental transfer of acetylcholine receptor (AChR) antibodies and usually recovers within two months of life.23
Clinical presentation is characterized by fatigability and fluctuating weakness of ocular, facial, limb, bulbar, and respiratory muscles. Antibodies against AChR, muscle-specific kinase (MuSK), and lipoprotein receptor-related protein 4 (LRP4) may participate in its pathogenesis.24
A study compared 114 Chinese juvenile MG patients with 207 young adults MG patients. AChR antibodies were found in 77%, 80%, and 81% of the 0–8 years, 8–18 years, and adult age ranges, respectively. Only one out of six patients that tested positive for MuSK and LRP4 antibodies was not an adult. Repetitive nerve stimulation during electromyography (EMG) may be useful for diagnosis, as well as the neostigmine test, but this should be performed only in a controlled and monitored setting, in the presence of staff who are skilled in cardiopulmonary resuscitation. Atropine should be immediately available.23
StrokeStroke may occur in children older than one month as frequently as 13/100,000 per year. Its incidence is higher in neonates (25 to 40/100,000) and is highest among premature infants (up to 100/100,000).25 The underlying mechanisms in pediatric patients include ischemic stroke, sinovenous thrombosis, and hemorrhagic stroke.26 Affected patients usually present with an acute-onset focal neurological deficit, e.g., hemiplegia, hemianesthesia, hemiataxia, unilateral facial palsy, and/or aphasia.27 Newborns may present with seizures.26
Of the children with arterial ischemic stroke, 20–30% will have recurrent strokes; therefore, early diagnosis is paramount.26 Appropriate investigation with neuroimaging should not be delayed.
Alternating hemiplegia of childhoodAlternating hemiplegia of childhood is a rare developmental disorder that is caused, in 75% of cases, by a mutation in the ATP1A3 gene of neuronal Na+/K+ ATPase.28 Its onset is before 1.5 years of age and its clinical features consist of episodic unilateral hemiplegia, dystonia, quadriplegia, and abnormal eye movements. Each episode lasts for minutes, hours, or longer, and frequently recurs. Affected children may have developmental delay and epilepsy.28 The calcium channel blocker flunarizine can reduce frequency, severity, and duration of dystonic and hemiplegic spells.28
Periodic paralysesPeriodic paralyses are associated with mutations in the sodium channel gene SCN4A, the calcium channel gene CACNA1S, or the potassium channel gene KCNJ2, and should not be confused with secondary causes of episodic paralysis, such as drug toxicity.29 Patients present with episodic attacks of flaccid muscle weakness, typically associated with hypo- or hyperkalemia. Episodes can be triggered by carbohydrate- or potassium-rich foods, or by rest after exercise. Treatment with acetazolamide may decrease the frequency and severity of episodes.29
Brainstem encephalitisBickerstaff's brainstem encephalitis (BBE) and Miller-Fisher syndrome form a continuous spectrum, both with ataxia and external ophthalmoplegia, but only those patients with BBE develop impaired consciousness.30 BBE is an uncommon disorder of unknown etiology; however, it is often associated with CNS cell surface antibodies, particularly anti-GQ1b antibodies.31 The most common initial symptoms are diplopia and gait disturbance; CSF albuminocytological dissociation is found in half the patients during the second week of illness, and facial or limb weakness may be observed.30
Spinal cord infarctionIt is rare in children, and may mimic other commons entities, such as ATM. Absence of CSF pleocytosis and a normal protein level help to differentiate between these two entities. It usually involves the anterior spinal artery. Risk factors are minor spinal trauma, parainfectious vasculopathies, and surgery.32 Children present with the typical spinal cord signs, such as a sensory level, bladder and/or bowel dysfunction, as well as paraplegia or tetraplegia.
Functional muscle weaknessAcute weakness may be caused by functional neurological disorder (FND), a condition that was formerly called hysteria and conversion disorder, and children as young as 5 years of age may be affected.33 In a British study of 204 children, whose ages ranged from 7 to 15 years, the most common presentation was weakness (63%) and abnormal movements (43%), and the most frequent antecedent stressor was bullying at school (81%).34 These authors estimated that the annual incidence of FND in the age range <15 years was 1.30/100,000. A sharp observer will detect a few incongruous findings during physical examination of a patient presenting with weakness due to FND6,33,35:
- -
monocular diplopia – the child complains of double vision which persists when one eye is closed. Nevertheless, monocular diplopia may also be caused by a refractive error, particularly astigmatism
- -
the child is unable to walk but can lift his/her legs against gravity when lying on a bed. The rest of the motor examination is normal.
- -
give-way weakness – sudden loss of tone after an initial good/normal strength response when a muscle is tested against resistance
- -
co-contraction – effortful contraction of one muscle and its agonist, resulting in almost no movement at the joint
- -
motor inconsistency – significant differences in motor performance under different testing conditions.
- -
Hoover sign – the examiner places a hand under the heel of the “good” leg and asks the patient to lift the weak leg against resistance. A patient with FND will not exert downward pressure with the “good” leg.
When a pediatric patient presents at the ER with a main complaint of acute motor deficit, it is worthwhile to pay close attention to a few key aspects of history and physical examination (Table 1). Figs. 1 and 2 suggest algorithms for investigating acute localized and diffuse weakness in the ER setting, respectively.

Algorithm for acute localized weakness investigation in the emergency setting.
AChR, acetylcholine receptor; CNS, central nervous system; EEG, electroencephalography; EMG, electromyography; GBS, Guillain–Barré syndrome; Hx, history; OPV, oral polio vaccine; PE, physical examination; RHS, Ramsay-Hunt syndrome; VAPP, vaccine-associated paralytic poliomyelitis. aA cause of pseudoparalysis.

Algorithm for acute diffuse weakness investigation in the emergency setting.
AChR, acetylcholine receptor; CK, serum creatine-kinase; CNS, central nervous system; CSF, cerebrospinal fluid; EEG, electroencephalography; EMG, electromyography; GBS, Guillain–Barré syndrome; Hx, history; IgG, immunoglobulin G; K+, serum potassium; MS, multiple sclerosis; NMO, neuromyelitis optica; PE, physical examination; PRES, posterior reversible encephalopathy syndrome; RHS, Ramsay-Hunt syndrome; T4, thyroxine; w/o, without.
aA cause of pseudoparalysis.
bMyelopathy signs include a sensory level, bladder and/or bowel dysfunction, and paraplegia or tetraplegia.
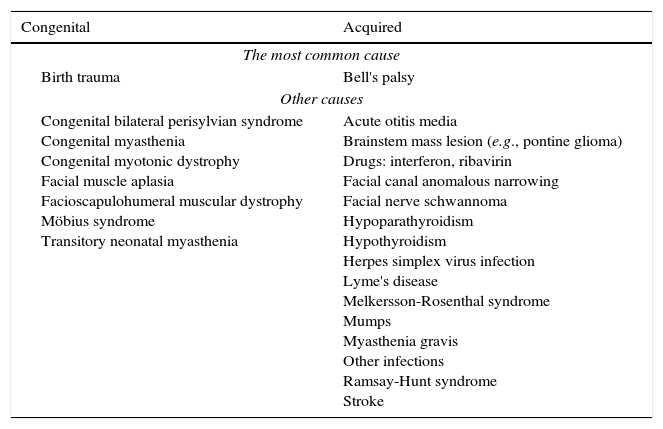
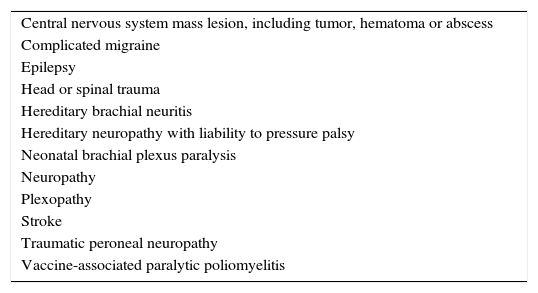
A localized deficit, such as facial palsy (Table 4)36 or monoplegia (Table 5),3 tends to have a smaller list of etiologies, while a more diffuse deficit, e.g., paraplegia, hemiplegia, or tetraplegia, will require consideration of many etiologies (Table 2). A word of caution is in order, as a thorough physical examination of a patient who complains of monoplegia may reveal inconspicuous weakness of an additional limb, so the patient is actually affected by hemiplegia or paraplegia.37
Etiologies of facial paralysis.
| Congenital | Acquired |
|---|---|
| The most common cause | |
| Birth trauma | Bell's palsy |
| Other causes | |
| Congenital bilateral perisylvian syndrome Congenital myasthenia Congenital myotonic dystrophy Facial muscle aplasia Facioscapulohumeral muscular dystrophy Möbius syndrome Transitory neonatal myasthenia | Acute otitis media Brainstem mass lesion (e.g., pontine glioma) Drugs: interferon, ribavirin Facial canal anomalous narrowing Facial nerve schwannoma Hypoparathyroidism Hypothyroidism Herpes simplex virus infection Lyme's disease Melkersson-Rosenthal syndrome Mumps Myasthenia gravis Other infections Ramsay-Hunt syndrome Stroke |
Source: Used with permission from Vasconcelos.36
Etiologies of monoplegia.
| Central nervous system mass lesion, including tumor, hematoma or abscess |
| Complicated migraine |
| Epilepsy |
| Head or spinal trauma |
| Hereditary brachial neuritis |
| Hereditary neuropathy with liability to pressure palsy |
| Neonatal brachial plexus paralysis |
| Neuropathy |
| Plexopathy |
| Stroke |
| Traumatic peroneal neuropathy |
| Vaccine-associated paralytic poliomyelitis |
Source: Adapted from Fenichel.3
When a patient presents with acute-onset diffuse weakness, the presence or absence of mental status changes, i.e., encephalopathy, will guide the diagnostic workup. In encephalopathic patients, CNS infection becomes a pressing hypothesis to be addressed by appropriate studies. Other entities need to be considered as well, such as seizures, mass lesions, dysfunction in other organ systems, trauma, and metabolic disturbances.
Great part of the neurologic examination deals with lesion localization, and the authors believe that a general pediatrician working in the ER should master the basic examining skills to ascertain whether an acute motor deficit stems from the brain, brainstem, spinal cord, neuromuscular junction, skeletal muscle, or peripheral nerves.
A large number of tests and procedures are available to investigate an acutely weak patient. As in similar situations, initial priority should be given to rule out life-threatening and treatable conditions. A stepwise, cost-effective, and timely workup is strongly recommended, as outlined in Figs. 1 and 2.
ConclusionsMost patients arriving in the ER with an acute motor deficit will benefit from a practical investigative approach and timely interventions based upon history, physical examination, and a few diagnostic tests, and will be able to eventually recover their well-being and full health.
Conflict of interestsThe authors declare no conflicts of interest.
Please cite this article as: Vasconcelos MM, Vasconcelos LG, Brito AR. Assessment of acute motor deficit in the pediatric emergency room. J Pediatr (Rio J). 2017;93:26–35.




