



To evaluate the clinical features associated with adrenocortical hormone overexpression and familial cancer profiling as potential markers for early detection of adrenocortical tumors in children from South and Southeast Brazil.
MethodsThe clinical manifestations and anthropometric measurements of 103 children diagnosed with adrenocortical tumors were analyzed.
ResultsBetween 1982 and 2011, 69 girls and 34 boys diagnosed with adrenocortical tumors were followed-up for a median time of 9.0 years (0–34 years). Signs of androgen overproduction alone (n=75) or associated with cortisol (n=18) were present in 90.3%. TP53 p.R337H mutation was found in 90.5% of patients. Stages I, II, III, and IV were observed in 45.6%, 27.2%, 19.4%, and 7.8% of patients, respectively. At diagnosis, there were no significant differences in height (p=0.92) and weight (p=0.22) among children with adrenocortical tumors, but children with virilization alone had significantly higher height-for-age Z-scores (0.92±1.4) than children with hypercortisolism alone or combined (−0.32±1,8; p=0.03). The five-year overall survival was 76.7% (SD±4.2). Patients with advanced-stage disease had a significantly worse prognosis than those with limited disease (p<0.001). During follow-up, ten of 55 p.R337H carrier parents developed cancer, whereas none of the 55 non-carriers did.
ConclusionsSigns of adrenocortical hormone overproduction appear early, even in cases with early-stage. These signs can be identified at the physical examination and anthropometric measurements. In southern Brazil, pediatric adrenocortical tumor is a sentinel cancer for detecting families with germline p.R337H mutation in TP53 gene.
Avaliar as manifestações clínicas da hiperexpressão de hormônios do córtex da adrenal e câncer familiar como marcadores para a detecção precoce de tumores adrenocorticais em crianças do Sul e Sudeste do Brasil.
Pacientes e métodosForam analisadas as manifestações clínicas e antropométricas de 103 crianças diagnosticadas com tumores adrenocorticais.
ResultadosEntre 1982 e 2011, 69 meninas e 34 meninos diagnosticados com tumores adrenocorticais foram acompanhados por um tempo mediano de nove anos (0-34). Ao diagnóstico, sinais de virilização isolada (n=75) ou associada ao cortisol (n=18) estavam presentes em 90,3% dos pacientes; a mutação do gene TP53 p.R337H foi identificada em 90,5% dos pacientes. Os pacientes foram classificados em estádio I (45,6%), II (27,2%), III (19,4%) e IV (7,8%). Ao diagnóstico, não houve diferença significativa para as medidas de altura (p=0,92) e de peso (p=0,22) entre as crianças com tumores adrenocorticais, mas crianças com virilização tiveram escore-Z mais elevado para a idade (0,92±1,4) do que aquelas com hipercortisolismo isolado ou combinado (−0,32±1,8; p=0,03). A sobrevida global de cinco anos foi de 76,7% (DP±4,2). Pacientes com estádios avançados tiveram pior prognóstico (p<0,001). Durante o seguimento, 10 dos 55 genitores portadores da p.R337H desenvolveram câncer, enquanto que nenhum caso ocorreu entre os 55 não portadores.
ConclusõesOs sinais de hiperprodução de hormônios adrenocorticais aparecem precocemente no desenvolvimento do tumor e podem ser identificados pelo exame físico e medidas antropométricas na consulta pediátrica de rotina. O tumor adrenocortical pediátrico é sentinela para a detecção de câncer em famílias que segregam a mutação germinativa p.R337H do gene TP53.
Adrenocortical tumors (ACT), common in southern and southeastern Brazil, constitute a spectrum of lesions with a behavior ranging from benign to malignant, affecting mainly children in the first 5 years of life. Early diagnosis and treatment are paramount for survival, since advanced cases can be highly aggressive, with high mortality.
The incidence of adrenocortical tumors (ACT) in the South and Southeast of Brazil reaches 3.4–4.2 cases per million in children under 15 years of age, representing an 18-fold higher frequency than that found in other regions of Brazil and worldwide.1–3 In over 90% of cases, they are associated with the germinal mutation of the TP53 tumor suppressor gene, located in the exon 10 of the short arm of chromosome 17, called p.R337H, which gives it a hereditary characteristic of predisposition to neoplasms that is, to date, unique to this population.4
The results of the neonatal screening studies, carried out in the metropolitan region of Campinas, state of São Paulo, and in the state of Paraná, Brazil, demonstrated that this mutation is present in approximately 1:300 newborns.5,6 The mutation was apparently introduced as a founding effect in this Brazilian region in the 18th century, during the Brazilian colonization period.7 The protein encoded by the TP53 gene is crucial to preserve genomic integrity, and mutations in this gene lead to a decrease in this protein activity and, consequently, to an increase in the likelihood of cancer in carrier individuals.8 Due to the strong association between TP53 mutation and ACT in children, this tumor is considered a sentinel for the detection of TP53 p.R337H mutation in Brazilian patients.9 Other genetic, epigenetic, and environmental factors collaborate to establish the risk in individuals that are susceptible mutation carriers.10
The clinical manifestations of ACTs are related to increased secretion of corticosteroids (mineralocorticoids, glucocorticoids, and sex hormones). The pediatrician can detect signs and symptoms in routine clinical and anthropometric assessments in childcare consultations, since approximately 90% of the tumors are secretory; those non-secretory are discovered at the clinical assessment or in ultrasound examinations for diagnostic investigation of increased abdominal volume, abdominal pain, or mass.11
In this article, the authors describe the main clinical, epidemiological, and biological characteristics of childhood ACT, with emphasis on family predisposition to cancer and strategies for early diagnosis, patient referral, and counseling to family members at risk of inheriting the mutation.
MethodsEligibilityBetween December 1982 and December 2014, 137 patients under 23 years of age were diagnosed with ACT at Centro Infantil Boldrini (CIB), a referral center in pediatric oncology in Campinas/SP. A total of 103 patients were eligible for the study, excluding patients lost at follow-up (n=10), perioperative deaths (n=4), and those with less than three years of diagnosis (n=20).
Longitudinal follow-upSince 2002, the study patients followed the same clinical and laboratory follow-up protocol for diagnosis of familial mutations, early recurrence detection, treatment sequelae, and occurrence of cancer in family members.
Growth and development measurements were performed at the six-monthly or annual consultations, considering clinical data and anthropometric measures (weight, height and body mass index [BMI]). The World Health Organization curves were used as reference.12,13 The Anthro Plus v.1.0.4 program (WHO AnthroPlus software, version 1.0.4, 2011) was used for the analysis of the Z-score of anthropometric measurements.
Genograms were also developed for three generations of the 55 families that participated in the family cancer study (Cyrillic v.2.1 program). All patients received genetic counseling and psychological support.
Laboratory and imaging diagnosisSerum levels of total and free testosterone, androstenedione, dehydroepiandrosterone, dehydroepiandrosterone sulfate, aldosterone, cortisol, adrenocorticotrophic hormone, progesterone, and estradiol were analyzed; chest X-ray and tomography, abdominal ultrasound, and magnetic resonance imaging (MRI) exams were performed.
At diagnosis, patients were classified according to the clinical manifestation of excess corticosteroid hormones secreted by the tumor: virilization syndrome (androgens), Cushing's syndrome (cortisol), mixed syndrome (virilization and Cushing's), and asymptomatic (non-secretory).
Tumor volume was measured at the imaging examination, and the surgical specimen was weighted. The pathological diagnosis followed the Weiss classification.14 To detect the mutation, molecular tests were performed using the polymerase chain reaction (PCR) method, followed by restriction enzyme and agarose gel.4
Clinical–surgical stagingPatients were staged according to Sandrini et al.1 as:
STAGE I – tumors <100g and <200cm3; completely resected; normal postoperative hormone levels;
STAGE II – tumors ≥100g or ≥200cm3; completely resected; normal postoperative hormone levels;
STAGE III – non-resectable tumors; macro- or microscopic residual disease; tumor rupture; high hormone levels after surgery; retroperitoneal lymph node involvement;
STAGE IV – distant metastases.
Treatment aspectsPatients with localized disease underwent total surgical resection of the tumor. Patients with tumors that were considered inoperable or had non-resectable distant metastases were submitted to biopsy for diagnostic confirmation and received chemotherapy (CT) prior to surgical resection of the primary tumor and metastases. Patients with advanced disease or intraoperative tumor rupture underwent intensive CT with a combination of the drugs cisplatin, etoposide, doxorubicin associated with mitotane (MTT) adrenocorticolytic agent (o, p’-DDD).
Statistical analysisThe following descriptive and association analyzes were performed: frequency tables for categorical variables; position and dispersion measurements for numerical variables; association between variables (chi-squared test or Fisher's exact test); for comparing numerical measures between groups (Mann–Whitney or Kruskal–Wallis test or ANOVA with rank transformation followed by Tukey's test); association between ACT and cancer (quasi-likelihood methods [WQLS] and generalized disequilibrium test [GDT]); survival analysis (Kaplan–Meier test) and Log-rank for comparison between curves. The level of significance was set at 5% and the Statistical Analysis System (SAS, software, version 9.4, NC, USA) was used for analysis.
Ethical aspectsThe study project of ACTs and the germline mutation in patients and their families was approved by the Research Ethics Committees of CIB and UNICAMP. The study subjects signed an informed consent form.
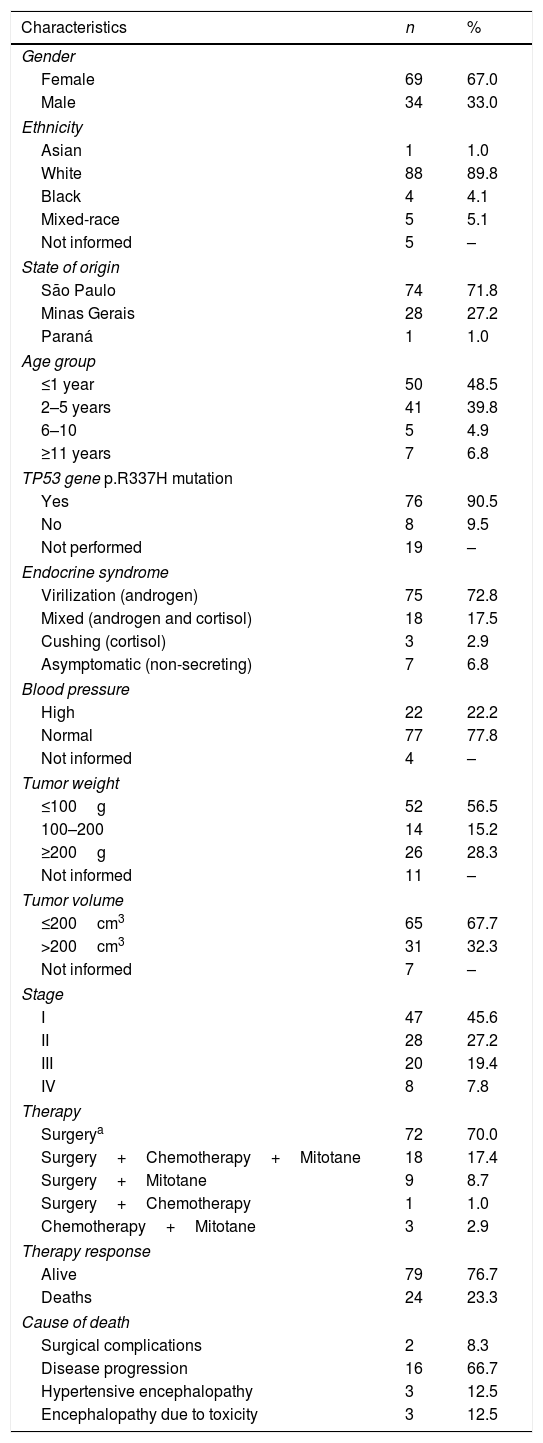
ResultsThe patients’ demographic, clinical, and evolution characteristics are shown in Table 1.
Demographic and clinical characteristics of 103 patients with adrenocortical tumors in childhood and adolescence.
| Characteristics | n | % |
|---|---|---|
| Gender | ||
| Female | 69 | 67.0 |
| Male | 34 | 33.0 |
| Ethnicity | ||
| Asian | 1 | 1.0 |
| White | 88 | 89.8 |
| Black | 4 | 4.1 |
| Mixed-race | 5 | 5.1 |
| Not informed | 5 | – |
| State of origin | ||
| São Paulo | 74 | 71.8 |
| Minas Gerais | 28 | 27.2 |
| Paraná | 1 | 1.0 |
| Age group | ||
| ≤1 year | 50 | 48.5 |
| 2–5 years | 41 | 39.8 |
| 6–10 | 5 | 4.9 |
| ≥11 years | 7 | 6.8 |
| TP53 gene p.R337H mutation | ||
| Yes | 76 | 90.5 |
| No | 8 | 9.5 |
| Not performed | 19 | – |
| Endocrine syndrome | ||
| Virilization (androgen) | 75 | 72.8 |
| Mixed (androgen and cortisol) | 18 | 17.5 |
| Cushing (cortisol) | 3 | 2.9 |
| Asymptomatic (non-secreting) | 7 | 6.8 |
| Blood pressure | ||
| High | 22 | 22.2 |
| Normal | 77 | 77.8 |
| Not informed | 4 | – |
| Tumor weight | ||
| ≤100g | 52 | 56.5 |
| 100–200 | 14 | 15.2 |
| ≥200g | 26 | 28.3 |
| Not informed | 11 | – |
| Tumor volume | ||
| ≤200cm3 | 65 | 67.7 |
| >200cm3 | 31 | 32.3 |
| Not informed | 7 | – |
| Stage | ||
| I | 47 | 45.6 |
| II | 28 | 27.2 |
| III | 20 | 19.4 |
| IV | 8 | 7.8 |
| Therapy | ||
| Surgerya | 72 | 70.0 |
| Surgery+Chemotherapy+Mitotane | 18 | 17.4 |
| Surgery+Mitotane | 9 | 8.7 |
| Surgery+Chemotherapy | 1 | 1.0 |
| Chemotherapy+Mitotane | 3 | 2.9 |
| Therapy response | ||
| Alive | 79 | 76.7 |
| Deaths | 24 | 23.3 |
| Cause of death | ||
| Surgical complications | 2 | 8.3 |
| Disease progression | 16 | 66.7 |
| Hypertensive encephalopathy | 3 | 12.5 |
| Encephalopathy due to toxicity | 3 | 12.5 |
The median time between the clinical manifestations and diagnosis was 4 months (0.3–36). The median follow-up time was 9 years (0–33.9). There was no significant difference regarding age at diagnosis between the genders (p=0.27). An association was observed between children under 5 years of age and stages I and II and between children older than 11 years and stages III and IV (p=0.02).
A correlation was observed between the symptoms at diagnosis and clinical staging for patients with virilization and stage I, patients with combined tumor and stages I and II, and asymptomatic patients and stages III and IV (p=0.01). Tumor weight greater than 200cm3 was correlated with advanced stages (III and IV; p<0.0001).
Seven patients had an association with congenital syndromes: multiple familial polyposis (2), pyelocalicial ectopy (1), NK cell immunodeficiency (1), double aortic arch (1), erythroderma variabilis (1), and Beckwith–Wiedemann syndrome (1). Of the 84 patients tested, 76 (90.4%) were carriers of the TP53 p.R337H germline mutation.
Two patients with the p.R337H mutation had synchronous neuroblastoma at diagnosis of ACT. There were two cases of second neoplasm, 22 and 10 years after the diagnosis of ACT; one non-smoker patient with lung adenocarcinoma and one patient with osteosarcoma, respectively.
Patients with localized disease were treated with surgery for complete tumor resection, and patients with advanced disease or recurrence received adjuvant therapy (Table 1).
The overall survival probability of the 103 patients, in a median follow-up period of 26.2 years (95% CI, 23.5–28.9), was 76.7% (SD±4.2; Fig. 1A). There was a significant difference in the overall survival probability between the stages (p<0.001): stage I, 95.7% (SD±0.3); II, 75% (SD±8.2); III, 55% (SD±11.1); and IV, 25% (SD±15.3; Fig. 1B).
 Overall survival; (B) stratified survival according to stage of the disease at diagnosis.")
Of the 179 first-degree relatives (parents, siblings, and children) of 55 probands, 175 were tested for the p.R337H mutation. Of these, 13 of the 81 carriers and one of the 94 non-carriers were diagnosed with cancer during a median follow-up period of 9.7 years (3–32; p=0.001). Among 33 carrier mothers, three developed breast cancer (at 41, 43, and 44 years of age);one developed adrenal carcinoma (at age 47 years); and one, leiomyosarcoma of the uterus (at age 61 years). Among the 22 carrier fathers, one developed a central nervous system tumor (at age 28 years); one, carcinoma of the larynx (at age 40 years); one, carcinoma of the esophagus (at age 46 years), one, lung adenocarcinoma (at age 51 years, non-smoker); and one, gastric cancer (at age 62 years). A proband's sister, who was a carrier, developed ACT at 1.4 years and one child, a carrier, developed neuroblastoma at 2.8 years; one sister, non-carrier, developed acute myeloid leukemia (at age 23 years). The median age of the parents at the cancer diagnosis was 45 years (28–62).
A family history of cancer was available in 50 of the 55 families in the mutation-segregating parental line (SPL) and 49 of the 55 in the mutation non-segregating parental line (NSPL); 47/50 (94%) and 27/49 (55%) had a history of cancer (p<0.001); multiple occurrences of cancer in first, second, and third-degree relatives were observed in 25 of the 47 (53.2%) SPL families and in four of 27 (14.8%) NSPL families (p=0.001).
The most common types of neoplasms were: breast cancer in 22/91 women, 61 gastrointestinal tumors, 17 central nervous system tumors, and 14 laryngeal tumors, in a total of 198 neoplasms detected in families in which the mutation is segregated.
In Fig. 2, a family genogram exemplifies cancer predisposition.
Anthropometry
The 64 surviving patients were eligible for anthropometric data assessment. At diagnosis, there was no significant difference for the mean and median age (p=0.53), weight Z-score (p=0.22), height (p=0.92), and BMI (p=0.27) between the female and male genders. Patients with signs and symptoms of androgen-secreting tumors had above-average height Z-scores for the same age and gender population. A statistical difference was observed for the height Z-score (p=0.03) between patients with virilization (0.92±1.4) and mixed syndromes (−0.32±1.8). After disease remission, in the follow-up consultations, anthropometric analyses indicated that there was no statistical difference between genders for BMI (p=0.67), but there was a difference regarding a higher height Z-score for the female gender (p=0.03); in the comparison between patients with virilization and combined syndrome, there was no significant difference for height Z-score (p=0.52) and for BMI Z-score (p=0.08; Fig. 3).
 Comparison of height (A) and BMI (B) Z-score between genders and between endocrine syndromes (virilization and mixed), at two moments of evaluation: at diagnosis and after therapy of 64 survivors (45 females and 19 males) of childhood ACT.")
The analysis of the height catch-down for patients with virilization showed a greater decrease in the Z-score for the male gender (p=0.03).
DiscussionIn this study, the authors confirmed that early diagnosis of children carrying the p.R337H mutation and ACT is associated with high curability without long-term health impairment. Surgery with complete tumor resection is sufficient to cure approximately 80% of children with small tumors. However, when the diagnosis is late, the tumors are more aggressive and the patients receive adjuvant treatment with intensive chemotherapy and mitotane, compromising prognosis and survival.
The results demonstrated that early signs and symptoms of ACT can be detected in childcare follow-up, as 86% of cases occur before 5 years of age and 93% of patients have somatic growth disorders or age-inappropriate sexual features.15,16 It should be noted that abnormal steroid secretion occurs early during tumor development and, therefore, subtle clinical manifestations are the first signs of the disease.
Height growth acceleration may be the first sign of high androgen secretion and rapid weight gain, of cortisol secretion; in this study, 75% of the children were above the 75th percentile for weight and height. For children with non-secretory tumors, the Z-score for weight and height were below average, probably due to secondary loss to tumor catabolism, as the diagnosis was attained later, when the disease was advanced.
Most cases present as early pseudopuberty with signs of virilization or as Cushing's syndrome, isolated or associated, easily mistaken for adrenal gland hyperplasia or central precocious puberty.17,18 The authors emphasize that, particularly in children from South and Southeast Brazil, the possibility of ACT should be the first hypothesis and imaging tests, ultrasound, or preferably MRI of the abdomen should be performed immediately, as the delay in diagnosis compromises prognosis and survival.19 Computed tomography is not indicated, due to the risk of exposure to irradiation. Due to the possible difficulties to perform the MRI, the authors suggest the abdominal ultrasound assessment, with special attention to the adrenal glands, and the measurement of hormones (androgens and cortisol), in addition to an investigation of the family history of cancer. The presence of virilization before 4 years of age, hypercortisolism before 10 years, disproportion between signs of virilization and testicular volume in boys, and presence of signs of two steroid chains are highly suggestive of ACT.16 In these cases and/or in the presence of adrenal mass, the child should be sent to the referral center as a matter of urgency.
Arterial hypertension was observed at diagnosis in approximately 22% of the patients and, in most cases, it was associated with cortisol production. Hypertension was more common in patients with Cushing and mixed syndrome; however, 7% of patients with virilization or non-secreting tumor had hypertension, probably due to compression of the renal artery by the tumor or increased aldosterone production.11
Other signs and symptoms related to secretory tumors that may be present are: pubarche or premature axillary hair (92%), clitoromegaly (92%), phallumegaly (91%), hirsutism (62%), increased volume or palpable mass in the abdomen (55%), acne (47%), facial plethora (42%), moon facies (35%), and increased voice pitch (32%); early thelarche is not a common sign, as estrogen-producing tumors are rare. Hypertension secondary to aldosterone secretion is generally a manifestation associated with virilization or hypercortisolism.11,20,21
In this study, non-secreting ACTs represented 6.8% of the cases and were diagnosed late through imaging tests, with advanced disease and reserved prognosis.
Prolonged exposure to androgens accelerates bone age and development, sometimes leading to precocious puberty requiring hormonal blockade.17,20 The present series included three girls in need of pubertal block at 6 and 7 years of age.
ACT prognosis is associated with disease staging at diagnosis. In this study, the survival probability for patients with localized disease, which could be surgically resected, was above 75%. Mitotane (o-p’DDD), an adrenocorticolytic agent, has been used as adjuvant to surgery, associated or not with conventional chemotherapy, but its true efficacy still needs to be demonstrated. Furthermore, mitotane has significant side effects.22 Radiation therapy is not recommended because, in over 90% of cases in southern Brazil, adrenal carcinoma is associated with TP53 mutations, which may predispose to secondary neoplasms in the irradiated area.21
The TP53 p.R337H mutation is associated with other neoplasms in childhood and adulthood. This study group demonstrated an association of p.R337H with other pediatric tumors in children treated at CIB, showing that, in addition to ACT, p.R337H is strongly associated with choroid plexus carcinoma (69%), osteosarcoma (7%) and neuroblastoma (8.4%) and in three out of 123 women with breast cancer in the Southeast region of Brazil.23–25
In all cases of patients with p.R337H mutation, the inheritance was confirmed by the presence of the mutation in one of the parents, corroborating the observation that, to date, in the South of Brazil, no cases of de novo mutations have been reported. In 2001, Ribeiro et al. described for the first time the missense mutation in exon 10 of the TP53 gene, originating the protein variant with substitution of an arginine by a histidine called p.R337H, and proved the association of this mutation with ACT.4 Other authors have reported the association of this mutation with families that have familial predisposition to cancer syndromes.26
In the prospective study of 55 families of children treated for ACT and carrying the mutation, the authors found 13 new cases of cancer among 81 tested first-degree relatives that were carriers and one case among 94 non-carrier relatives. In the literature, other non-p.R337H mutations of TP53 are associated with familial cancer syndrome, such as the classic Li-Fraumeni syndrome and its Li-Fraumeni-like variants (OMIM # 151623), but with a spectrum of different tumors (sarcomas, breast carcinoma, central nervous system tumors, and leukemia) than those found in the family members of this study and different age at cancer occurrence in younger individuals.27–29
The authors advise including the family history of cancer in the anamnesis and the three-generation genogram to alert for the presence of the mutation, as, because it is hereditary, individuals in the segregating line are at risk of developing cancer. The presence of cancer in more than one first or second-degree relative, or multiple tumors in the same individual or cancer in young individuals indicate family predisposition.
The authors of this study demonstrated the high frequency of ACT in a reference center in Southeast Brazil, which was associated with the hereditary TP53 p.R337H mutation in more than 90% of the cases. Most cases can be detected early through signs and symptoms of elevated corticosteroid secretion; and survival is associated with early diagnosis and localized disease. ACT diagnosis in a child represents an alert for family predisposition to cancer.
The pediatrician, in primary health care, is the fundamental link between diagnostic suspicion and appropriate treatment. It is up to the referral centers to develop therapeutic protocol strategies that provide the pediatrician with new forms of prevention and predictive measures for children and families at risk of developing cancer.
FundingProject coordinated by Prof. José Andrés Yunes with grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Number 401991/2010 and Fundação Capes, Ministry of Education. PROCAD N. 247/2007, for molecular biology analyses.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank Gisele Miniussi de Assis, data manager of the Study Program of Adrenocortical Tumors in Childhood and the Research protocol ARAR0332 of the Children's Oncology Group, developed at Centro Infantil Boldrini and UNICAMP.
The authors would also like to thank Cleide Aparecida Moreira Silva, statistician of the Statistics Department of Faculdade de Ciências Médicas da UNICAMP.
Please cite this article as: Mastellaro MJ, Ribeiro RC, Oliveira-Filho AG, Seidinger AL, Cardinalli IA, Miranda EC, et al. Adrenocortical tumors associated with the TP53 p.R337H germline mutation can be identified during child-care consultations. J Pediatr (Rio J). 2018;94:432–9.




