

The prevalence of non‐alcoholic fatty liver disease in children has risen significantly, owing to the worldwide childhood obesity epidemic in the last two decades. Non‐alcoholic fatty liver disease is closely linked to sedentary lifestyle, increased body mass index, and visceral adiposity. In addition, individual genetic variations also have a role in the development and progression of non‐alcoholic fatty liver disease. The aim of this study was to investigate the gene polymorphisms of MCP‐1 (‐2518 A/G) (rs1024611), CCR‐2 (190 G/A) (rs1799864), ABCA1 (883 G/A) (rs4149313), and IL‐17A (‐197 G/A) (rs2275913) in obese Turkish children with non‐alcoholic fatty liver disease.
MethodsThe study recruited 186 obese children aged 10-17 years, including 101 children with non‐alcoholic fatty liver disease and 85 children without non‐alcoholic fatty liver disease. Anthropometric measurements, insulin resistance, a liver panel, a lipid profile, liver ultrasound examination, and genotyping of the four variants were performed.
ResultsNo difference was found between the groups in respect to age and gender, body mass index, waist/hip ratio, or body fat ratio. In addition to the elevated ALT levels, AST and GGT levels were found significantly higher in the non‐alcoholic fatty liver disease group compared to the non non‐alcoholic fatty liver disease group (p<0.05). The A‐allele of IL‐17A (‐197 G/A) (rs2275913) was associated with non‐alcoholic fatty liver disease (odds ratio [OR] 2.05, 95% confidence interval: 1.12‐3.77, p=0.02).
ConclusionsThe findings of this study suggest that there may be an association between IL‐17A (‐197 G/A) (rs2275913) polymorphism and non‐alcoholic fatty liver disease development in obese Turkish children.
A prevalência de doença hepática gordurosa não alcoólica em crianças aumentou significativamente devido à epidemia de obesidade infantil em todo o mundo nas últimas duas décadas. A doença hepática gordurosa não alcoólica está intimamente ligada ao estilo de vida sedentário, ao aumento do índice de massa corporal e à adiposidade visceral. Além disso, variações genéticas individuais também têm um papel no desenvolvimento e na progressão da doença hepática gordurosa não alcoólica. O objetivo deste estudo foi investigar os polimorfismos genéticos MCP‐1 (‐2518 A/G) (rs1024611), CCR‐2 (190 G/A) (rs1799864), ABCA1 (883 G/A) (rs4149313) e IL‐17A (‐197 G/A) (rs2275913) em crianças turcas obesas com doença hepática gordurosa não alcoólica.
MétodosO estudo recrutou 186 crianças obesas entre 10 e 17 anos, inclusive 101 crianças com doença hepática gordurosa não alcoólica e 85 crianças sem doença hepática gordurosa não alcoólica. Medidas antropométricas, resistência à insulina, painel hepático, perfil lipídico, exame ultrassonográfico do fígado e genotipagem de quatro variantes foram feitos.
ResultadosNenhuma diferença foi encontrada entre os grupos em relação à idade e sexo, índice de massa corporal, relação cintura/quadril ou proporção de gordura corporal. Além dos níveis elevados de ALT, os níveis de AST e GGT foram significativamente maiores no grupo doença hepática gordurosa não alcoólica em comparação com o grupo não doença hepática gordurosa não alcoólica (p < 0,05). O alelo A de IL‐17A (‐197 G/A) (rs2275913) foi associado à doença hepática gordurosa não alcoólica (odds ratio [OR] 2,05, intervalo de confiança de 95%: 1,12‐3,77, p=0,02).
ConclusõesOs achados deste estudo sugerem que pode haver uma associação entre o polimorfismo IL‐17A (‐197 G/A) (rs2275913) e o desenvolvimento da doença hepática gordurosa não alcoólica em crianças turcas obesas.
A doença hepática gordurosa não alcoólica (DHGNA) é uma doença hepática crônica causada pelo acúmulo excessivo de gordura no fígado, sem história de consumo crônico de álcool, doenças metabólicas do fígado (doença de Wilson) ou doenças hepáticas congênitas, virais ou autoimunes.1 A DHGNA está incluída em um amplo espectro de doenças hepáticas, variam de simples esteatose a esteato‐hepatite, fibrose e até cirrose.1 A prevalência de DHGNA aumentou significativamente, devido à epidemia mundial de obesidade infantil nas duas últimas décadas.2 A DHGNA está intimamente ligada a um estilo de vida sedentário, aumento do índice de massa corporal (IMC) e adiposidade visceral em crianças, resultante do consumo excessivo de calorias.3 Vários estudos recentes relatam que, além dos fatores de risco ambientais, as variações genéticas individuais também podem contribuir para o desenvolvimento e a progressão da DHGNA.4
Vários genes candidatos foram implicados na patogênese da DHGNA, inclusive genes envolvidos no metabolismo lipídico hepático, sensibilidade à insulina e geração de espécies oxidantes reativas ou citocinas.5 Polimorfismos de nucleotídeo único em genes envolvidos no metabolismo lipídico (domínio de fosfolipase tipo‐patatina‐3 e lipina 1), estresse oxidativo (superóxido dismutase 2), sinalização de insulina (substrato receptor de insulina‐1) e fibrogênese (fator Kruppel‐like 6) foram associados à DHGNA em crianças.2 Além desses polimorfismos bem conhecidos, polimorfismos em genes que codificam proteínas envolvidas na patogênese da DHGNA também podem estar associados ao desenvolvimento da doença.
A proteína quimioatrativa de monócitos 1 (MCP‐1), também chamada de quimiocina ligante 2 (CCL2), um forte fator quimiotático, desempenha um papel na ativação de monócitos, macrófagos e células T nas fases aguda e crônica da inflamação.6 O polimorfismo 2518 A / G no gene da MCP‐1 afeta o nível de expressão da MCP‐1 em resposta a estímulos inflamatórios e sabe‐se que isso está associado a diabetes mellitus e várias doenças autoimunes.7,8 A MCP‐1 exerce um efeito quimiotático sobre monócitos e células T através do receptor de quimiocinas 2 (CCR2).9 O polimorfismo 190 G/A no gene CCR também já foi associado ao desenvolvimento de DHGNA.10
O transportador‐1 de cassete de ligação de ATP (ABCA1) é um membro da família de transportadores de membrana de cassete de ligação de ATP (ABC). A expressão de ABCA1 resultou em aumento do efluxo de colesterol e diminuição do conteúdo lipídico hepático.11 O estresse inflamatório aumenta o acúmulo de colesterol nos hepatócitos e inibe a expressão de ABCA1.12 Além disso, foi relatado que a diminuição da expressão hepática de ABCA1 pode causar esteato‐hepatite em pacientes adultos com obesidade mórbida.11 A interleucina‐17 (IL‐17) é uma molécula pró‐inflamatória produzida por células T‐helper (Th) 17. Ela age como mediador da resposta imune contra patógenos bacterianos e fúngicos extracelulares e está envolvida no desenvolvimento de doenças inflamatórias e autoimunes.13 Também tem sido sugerido que a IL‐17 tem um papel crítico no desenvolvimento de várias doenças hepáticas, como hepatite viral crônica, doenças autoimunes e doenças hepáticas alcoólicas.14–16 Foi demonstrado que a IL‐17A (‐197 G / A) afeta o desenvolvimento da colite ulcerativa e da artrite reumatoide.17
O objetivo do nosso estudo foi investigar a associação entre DHGNA e polimorfismos em genes que codificam proteínas cujo papel na patogênese da DHGNA tem sido sugerido. Que seja de nosso conhecimento, a relação entre os quatro polimorfismos de nucleotídeo único e a DHGNA não foi estudada anteriormente.
MétodosO estudo incluiu pacientes turcos obesos entre 10 e 17 anos, que se apresentaram na Clínica Pediátrica de Gastroenterologia, Hepatologia e Nutrição e Endocrinologia Pediátrica do Kanuni Training and Research Hospital entre junho de 2015 e julho de 2016. O paciente era considerado obeso se o escore z do IMC fosse ≥ 2 para idade e sexo.18 Os 186 pacientes incluídos no estudo foram separados em dois grupos. O grupo DHGNA (n=101) incluiu pacientes com diagnóstico de DHGNA, níveis de alanina aminotransferase (ALT) acima do dobro do limite superior da normalidade (sexo masculino > 50 U/L, sexo feminino > 44 U/L) e doença hepática gordurosa detectada por ultrassonografia (USG).19 Pacientes com outras causas de doença hepática gordurosa, como a doença de Wilson, deficiência de α‐1 antitripsina, hepatite autoimune e hepatite viral, foram excluídos. O grupo sem DHGNA (n=85) incluiu pacientes obesos sem DHGNA (níveis normais de ALT e imagem USG do fígado normal), com medidas antropométricas, resistência à insulina e perfil lipídico semelhantes aos do grupo DHGNA. Nenhum dos pacientes no estudo foi submetido a biópsia hepática. Pacientes com doenças genéticas, endócrinas ou metabólicas que podem causar obesidade não foram incluídos no estudo. Além disso, nenhum dos pacientes apresentava doenças pró‐inflamatórias comórbidas, como doença inflamatória intestinal.
O estudo foi feito após a aprovação do comitê de ética local (URL do Registro: 2015/27 Identifier: Trabzon Kanuni Training and Research Hospital Clinical Research Ethics Committee) e da obtenção do consentimento informado dos pais, de acordo com a Declaração de Helsinque.
Todos os participantes foram submetidos a exame físico. A altura foi medida até o centímetro mais próximo com um estadiômetro Harpenden (Holtain Limited, Crymych, Dyfed, País de Gales). O peso foi medido sem roupas até o 0,1kg mais próximo, com uma balança calibrada. O índice de massa corporal (IMC) foi calculado através da fórmula peso (kg)/altura (m2). Os escores Z do IMC foram calculados com os valores de referência para crianças turcas.20 A proporção de gordura corporal foi calculada pelo método de bioimpedância elétrica (BIA) em um dispositivo Tanita BC 418 (Tanita Corporation, Tóquio, Japão). As medidas da cintura e da circunferência do quadril foram obtidas com uma fita métrica inelástica, enquanto o paciente permanecia de pé com os pés juntos (12‐15cm) e os braços ao lado do corpo. A relação cintura‐quadril foi calculada dividindo‐se a medida da cintura pela medida do quadril.
Após 10 horas de jejum, amostras de sangue venoso periférico foram obtidas para determinar os níveis de insulina (Beckman Coulter DXI 800, Califórnia, EUA), lipoproteína de alta densidade (HDL), lipoproteína de baixa densidade (LDL), triglicérides, colesterol total, alanina aminotransferase (ALT), aspartato aminotransferase (AST), gama glutamiltransferase (GGT) e glicose (Beckman Coulter AU5821, Califórnia, EUA). O modelo homeostático para resistência insulínica (HOMA‐IR) foi calculado pela fórmula glicose × insulina (μU/mL)/405.21
Todos os exames foram feitos por dois radiologistas com mais de 10 anos de experiência em imagiologia abdominal pediátrica e esteatose hepática e os resultados foram determinados por consenso. Os radiologistas estavam cegos para os achados clínicos e resultados laboratoriais dos pacientes. A máquina de ultrassom usada foi a Aplio 500 (Toshiba Medical Systems, Otawara, Japão) com transdutores lineares de 4 a 9MHz. Os pacientes foram avaliados em decúbito dorsal com o braço direito em abdução máxima. Os pacientes foram submetidos ao exame após um jejum de 4 horas. Imagens longitudinais do lobo hepático direito e do rim ipsilateral foram obtidas, inclusive os planos hepáticos sagitais. A graduação semiquantitativa de alterações gordurosas foi feita com os achados da USG de fígado gorduroso, perda de definição das margens vasculares e atenuação profunda com contraste fígado‐rim.22
O DNA foi isolado do sangue periférico com o dispositivo automático QuickGene. Áreas alvo foram amplificadas por PCR com os pares primários: para MCP1 (2518 A/G) polimorfismo, F: 5’ CTTTCCCTTGTGTGTCCCC 3’, R: 5’ TTACTCCTTTTCTCCCCAACC 3’; para CCR‐2 rs1799864 polimorfismo, F: 5’ ATTTCCCCAGTACATCCACAAC 3’, R: 5’ CCCACAATGGGAGAGTAATAAG 3’; para ABCA1rs4149313 polimorfismo, F: 5’ GAGAAGAGCCACCCTGGTTCCAACCAGAAGAGGAT 3’, R: 5’ AGAAAGGCAGGAGACAT CGCTT 3’; e para IL‐17A rs2275913 polimorfismo, F: 5’ AACAAGTAAGAATGAAAAGAGGACATGGT 3’, R: 5’ CCCCCAATGAGGTCATAGAAGAATC 3’.
O seccionamento foi feito com enzimas de restrição Pvull (Vivantis) para MCP1 (‐2518 A/G) na atribuição do genótipo, enzimas de restrição FokL (Vivantis) para CCR‐2 rs1799864 na atribuição do genótipo, enzimas de restrição Econl (Vivantis) para ABCA1 rs4149313 na atribuição do genótipo e enzimas de restrição BstENI para IL‐17A rs2275913 na atribuição do genótipo. Após a separação por eletroforese em gel de agarose a 2%, a análise foi feita de acordo com o tamanho do produto. Para o controle, a precisão foi confirmada por análise de sequência aleatória de 10% em ambos os grupos.
Os dados foram avaliados com o software estatístico SPSS 13.0 (SPSS Inc., Chicago, IL, EUA). Os dados descritivos foram apresentados como média ± desvio‐padrão (DP). A análise dos resultados foi feita com a distribuição percentual para dados qualitativos e mediana do intervalo interquartil (IIQ) ou média (desvio‐padrão) para dados quantitativos. Para a comparação entre dois grupos, o teste t de amostras independentes pareadas foi usado para variáveis que apresentavam distribuição normal, enquanto o teste U de Mann Whitney foi usado para aquelas sem distribuição normal. No caso de mais de dois grupos, foi usado o teste Anova para variáveis com distribuição normal e o teste de Kruskal‐Wallis para aquelas sem distribuição normal. O teste do qui‐quadrado foi usado para comparar variáveis categóricas. A associação genotípica e odds ratio (OR) com intervalo de confiança de 95% (IC95%) foram estimados por análise de regressão logística binária. Em todos os casos, valores de p inferiores a 0,05 foram considerados estatisticamente significativos. A análise de potência foi feita com aproximação normal com o método de correção de continuidade do programa Open Epi, versão 3.01 (OpenEpi: Open Source Epidemiologic Statistics for Public Health, GA, EUA).
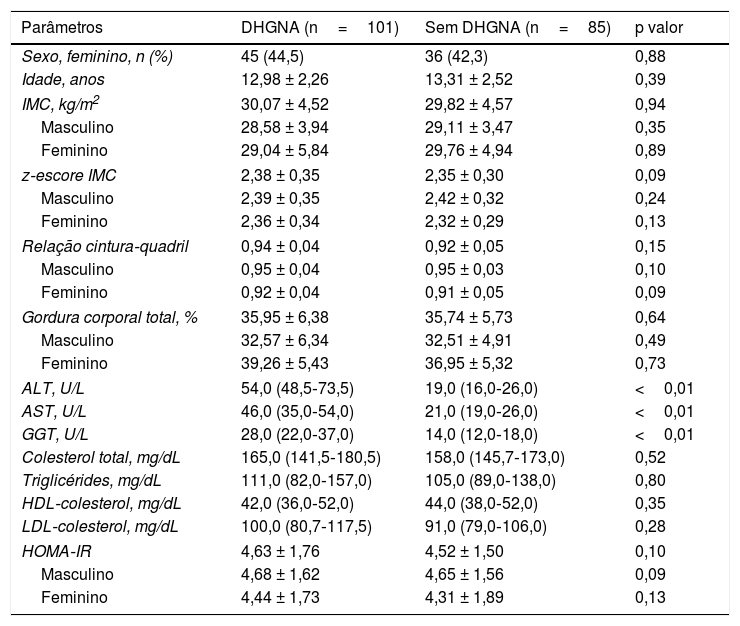
ResultadosAs características demográficas, medidas antropométricas e dados laboratoriais dos pacientes são mostrados na tabela 1. Não foram encontradas diferenças entre os grupos em termos de idade e sexo, IMC, relação cintura/quadril ou proporção de gordura corporal. Além da ALT, as concentrações de AST e GGT também foram significativamente maiores no grupo DHGNA do que no grupo sem DHGNA (p < 0,05).
Características dos grupos
| Parâmetros | DHGNA (n=101) | Sem DHGNA (n=85) | p valor |
|---|---|---|---|
| Sexo, feminino, n (%) | 45 (44,5) | 36 (42,3) | 0,88 |
| Idade, anos | 12,98 ± 2,26 | 13,31 ± 2,52 | 0,39 |
| IMC, kg/m2 | 30,07 ± 4,52 | 29,82 ± 4,57 | 0,94 |
| Masculino | 28,58 ± 3,94 | 29,11 ± 3,47 | 0,35 |
| Feminino | 29,04 ± 5,84 | 29,76 ± 4,94 | 0,89 |
| z‐escore IMC | 2,38 ± 0,35 | 2,35 ± 0,30 | 0,09 |
| Masculino | 2,39 ± 0,35 | 2,42 ± 0,32 | 0,24 |
| Feminino | 2,36 ± 0,34 | 2,32 ± 0,29 | 0,13 |
| Relação cintura‐quadril | 0,94 ± 0,04 | 0,92 ± 0,05 | 0,15 |
| Masculino | 0,95 ± 0,04 | 0,95 ± 0,03 | 0,10 |
| Feminino | 0,92 ± 0,04 | 0,91 ± 0,05 | 0,09 |
| Gordura corporal total, % | 35,95 ± 6,38 | 35,74 ± 5,73 | 0,64 |
| Masculino | 32,57 ± 6,34 | 32,51 ± 4,91 | 0,49 |
| Feminino | 39,26 ± 5,43 | 36,95 ± 5,32 | 0,73 |
| ALT, U/L | 54,0 (48,5‐73,5) | 19,0 (16,0‐26,0) | <0,01 |
| AST, U/L | 46,0 (35,0‐54,0) | 21,0 (19,0‐26,0) | <0,01 |
| GGT, U/L | 28,0 (22,0‐37,0) | 14,0 (12,0‐18,0) | <0,01 |
| Colesterol total, mg/dL | 165,0 (141,5‐180,5) | 158,0 (145,7‐173,0) | 0,52 |
| Triglicérides, mg/dL | 111,0 (82,0‐157,0) | 105,0 (89,0‐138,0) | 0,80 |
| HDL‐colesterol, mg/dL | 42,0 (36,0‐52,0) | 44,0 (38,0‐52,0) | 0,35 |
| LDL‐colesterol, mg/dL | 100,0 (80,7‐117,5) | 91,0 (79,0‐106,0) | 0,28 |
| HOMA‐IR | 4,63 ± 1,76 | 4,52 ± 1,50 | 0,10 |
| Masculino | 4,68 ± 1,62 | 4,65 ± 1,56 | 0,09 |
| Feminino | 4,44 ± 1,73 | 4,31 ± 1,89 | 0,13 |
ALT, alanina aminotransferase; AST, aspartato aminotransferase; DHGNA, doença hepática gordurosa não alcoólica; GGT, gama glutamiltransferase; HOMA‐IR, avaliação do modelo homeostático para resistência insulínica; IMC, índice de massa corporal.
Os valores são expressos como mediana (percentis 25 a 75) ou média ± desvio‐padrão, conforme apropriado.
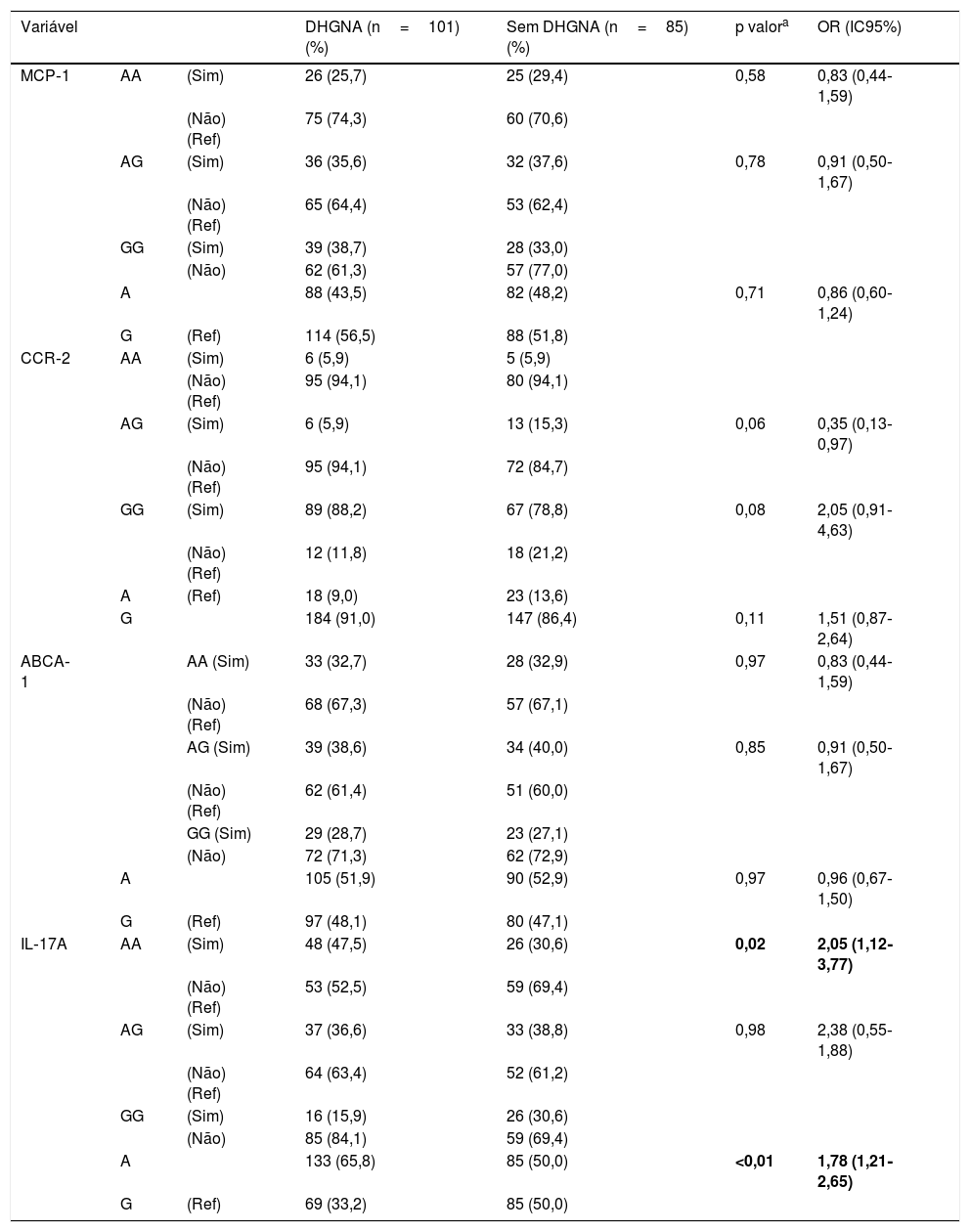
Não foram observadas diferenças entre os grupos em termos de genótipo e frequência alélica para os polimorfismos MCP‐1 rs1024611, CCR‐2 rs1799864 e ABCA1 rs4149313 (tabela 2). Dentre as quatro variantes, apenas o IL‐17A rs2275913 foi associado à DHGNA. A porcentagem de genótipos IL‐17A rs2275913 AA foi significativamente maior no grupo DHGNA do que no grupo sem DHGNA (p=0,02, OR: 2,05, IC95%: 1,12‐3,77). A frequência do alelo A de IL‐17A rs2275913 foi significativamente maior no grupo DHGNA do que no grupo sem DHGNA (p=< 0,01). O poder do estudo foi calculado em 53%. Para esse poder (erro alfa: 0,05 e Df: 1), o tamanho do efeito foi calculado em 0,15.
Frequência genotípica e alélica dos polimorfismos MCP‐1 rs1024611, CCR‐2 rs1799864, ABCA1 rs4149313, IL‐17A rs2275913 em pacientes e análise de regressão logística de fatores preditivos para DHGNA
| Variável | DHGNA (n=101) (%) | Sem DHGNA (n=85) (%) | p valora | OR (IC95%) | ||
|---|---|---|---|---|---|---|
| MCP‐1 | AA | (Sim) | 26 (25,7) | 25 (29,4) | 0,58 | 0,83 (0,44‐1,59) |
| (Não) (Ref) | 75 (74,3) | 60 (70,6) | ||||
| AG | (Sim) | 36 (35,6) | 32 (37,6) | 0,78 | 0,91 (0,50‐1,67) | |
| (Não) (Ref) | 65 (64,4) | 53 (62,4) | ||||
| GG | (Sim) | 39 (38,7) | 28 (33,0) | |||
| (Não) | 62 (61,3) | 57 (77,0) | ||||
| A | 88 (43,5) | 82 (48,2) | 0,71 | 0,86 (0,60‐1,24) | ||
| G | (Ref) | 114 (56,5) | 88 (51,8) | |||
| CCR‐2 | AA | (Sim) | 6 (5,9) | 5 (5,9) | ||
| (Não) (Ref) | 95 (94,1) | 80 (94,1) | ||||
| AG | (Sim) | 6 (5,9) | 13 (15,3) | 0,06 | 0,35 (0,13‐0,97) | |
| (Não) (Ref) | 95 (94,1) | 72 (84,7) | ||||
| GG | (Sim) | 89 (88,2) | 67 (78,8) | 0,08 | 2,05 (0,91‐4,63) | |
| (Não) (Ref) | 12 (11,8) | 18 (21,2) | ||||
| A | (Ref) | 18 (9,0) | 23 (13,6) | |||
| G | 184 (91,0) | 147 (86,4) | 0,11 | 1,51 (0,87‐2,64) | ||
| ABCA‐1 | AA (Sim) | 33 (32,7) | 28 (32,9) | 0,97 | 0,83 (0,44‐1,59) | |
| (Não) (Ref) | 68 (67,3) | 57 (67,1) | ||||
| AG (Sim) | 39 (38,6) | 34 (40,0) | 0,85 | 0,91 (0,50‐1,67) | ||
| (Não) (Ref) | 62 (61,4) | 51 (60,0) | ||||
| GG (Sim) | 29 (28,7) | 23 (27,1) | ||||
| (Não) | 72 (71,3) | 62 (72,9) | ||||
| A | 105 (51,9) | 90 (52,9) | 0,97 | 0,96 (0,67‐1,50) | ||
| G | (Ref) | 97 (48,1) | 80 (47,1) | |||
| IL‐17A | AA | (Sim) | 48 (47,5) | 26 (30,6) | 0,02 | 2,05 (1,12‐3,77) |
| (Não) (Ref) | 53 (52,5) | 59 (69,4) | ||||
| AG | (Sim) | 37 (36,6) | 33 (38,8) | 0,98 | 2,38 (0,55‐1,88) | |
| (Não) (Ref) | 64 (63,4) | 52 (61,2) | ||||
| GG | (Sim) | 16 (15,9) | 26 (30,6) | |||
| (Não) | 85 (84,1) | 59 (69,4) | ||||
| A | 133 (65,8) | 85 (50,0) | <0,01 | 1,78 (1,21‐2,65) | ||
| G | (Ref) | 69 (33,2) | 85 (50,0) | |||
DHGNA, doença hepática gordurosa não alcoólica; IC, intervalo de confiança; OR, odds ratio; Ref, referência.
Comparado com o grupo de genótipos de referência, as diferenças nas frequências genotípicas foram analisadas com o teste do qui‐quadrado. P <0,05 foi considerado estatisticamente significante. Todos os quatro polimorfismos foram incluídos na análise de regressão logística multivariada ao mesmo tempo.
Modelos: MCP‐1 AA, genótipo AG e alelo A, CCR‐2AG, genótipo GG e alelo G, ABCA‐1 AA, genótipo AG e alelo A, IL‐17A AA, genótipo AG e alelo A; Variável dependente: presença de DHGNA.
Os valores em negrito referem‐se a valores de p <0,05.
A análise de regressão logística univariada mostrou que pacientes com o genótipo IL‐17A rs2275913 AA tinham três vezes (IC 95%: 1,37‐6,58; p ≤ 0,01) mais chance de ter DHGNA do que pacientes com o genótipo GG (tabela 3).
Análise de regressão logística univariada de fatores preditivos para DHGNA
| Variável | Estimativa de coeficiente | EP | OR (IC95%) | p valor |
|---|---|---|---|---|
| IL‐17A rs2275913 AA genótipoa | 1,10 | 0,40 | 3,00 (1,37‐6,58) | <0,01 |
| IL‐17A rs2275913 AG genótipo | 0,60 | 0,40 | 1,82 (0,83‐3,97) | 0,13 |
DHGNA, doença hepática gordurosa não alcoólica; IC, intervalo de confiança; OR, odds ratio.
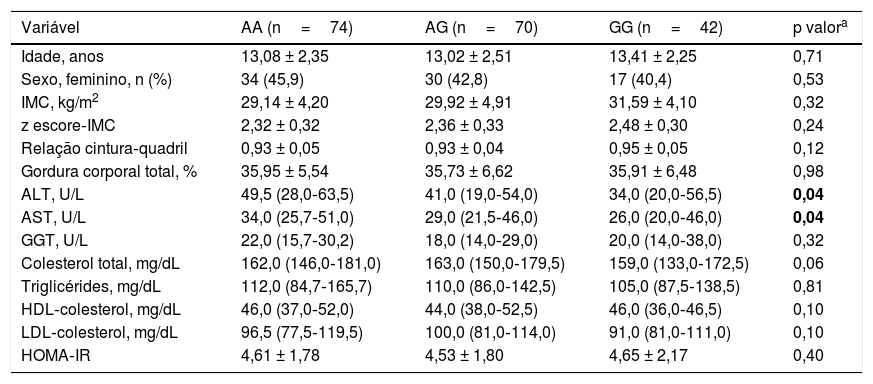
As características clínicas e bioquímicas dos grupos em relação ao genótipo IL‐17A rs2275913 são mostradas na tabela 4. Houve diferenças significativas nas concentrações de AST e ALT entre os três grupos (p=0,04 e p=0,04, respectivamente). As concentrações séricas de ALT e AST foram maiores no genótipo AA e menores no genótipo GG.
Características básicas dos indivíduos nos genótipos de IL‐17A rs2275913
| Variável | AA (n=74) | AG (n=70) | GG (n=42) | p valora |
|---|---|---|---|---|
| Idade, anos | 13,08 ± 2,35 | 13,02 ± 2,51 | 13,41 ± 2,25 | 0,71 |
| Sexo, feminino, n (%) | 34 (45,9) | 30 (42,8) | 17 (40,4) | 0,53 |
| IMC, kg/m2 | 29,14 ± 4,20 | 29,92 ± 4,91 | 31,59 ± 4,10 | 0,32 |
| z escore‐IMC | 2,32 ± 0,32 | 2,36 ± 0,33 | 2,48 ± 0,30 | 0,24 |
| Relação cintura‐quadril | 0,93 ± 0,05 | 0,93 ± 0,04 | 0,95 ± 0,05 | 0,12 |
| Gordura corporal total, % | 35,95 ± 5,54 | 35,73 ± 6,62 | 35,91 ± 6,48 | 0,98 |
| ALT, U/L | 49,5 (28,0‐63,5) | 41,0 (19,0‐54,0) | 34,0 (20,0‐56,5) | 0,04 |
| AST, U/L | 34,0 (25,7‐51,0) | 29,0 (21,5‐46,0) | 26,0 (20,0‐46,0) | 0,04 |
| GGT, U/L | 22,0 (15,7‐30,2) | 18,0 (14,0‐29,0) | 20,0 (14,0‐38,0) | 0,32 |
| Colesterol total, mg/dL | 162,0 (146,0‐181,0) | 163,0 (150,0‐179,5) | 159,0 (133,0‐172,5) | 0,06 |
| Triglicérides, mg/dL | 112,0 (84,7‐165,7) | 110,0 (86,0‐142,5) | 105,0 (87,5‐138,5) | 0,81 |
| HDL‐colesterol, mg/dL | 46,0 (37,0‐52,0) | 44,0 (38,0‐52,5) | 46,0 (36,0‐46,5) | 0,10 |
| LDL‐colesterol, mg/dL | 96,5 (77,5‐119,5) | 100,0 (81,0‐114,0) | 91,0 (81,0‐111,0) | 0,10 |
| HOMA‐IR | 4,61 ± 1,78 | 4,53 ± 1,80 | 4,65 ± 2,17 | 0,40 |
ALT, alanina aminotransferase; AST, aspartato aminotransferase; GGT, gama glutamiltransferase; HOMA‐IR, avaliação do modelo homeostático para resistência insulínica; IMC, índice de massa corporal.
No presente estudo, avaliamos o efeito de quatro polimorfismos no desenvolvimento de DHGNA em crianças obesas. Encontramos o genótipo AA do gene IL‐17A com maior frequência em crianças obesas com DHGNA do que em crianças obesas sem DHGNA. Entretanto, não houve diferenças significativas nos polimorfismos dos genes MCP‐1 rs1024611, CCR‐2 rs1799864 e ABCA1 rs4149313 em crianças turcas obesas com e sem DHGNA, sugeriu‐se que esses polimorfismos não tiveram papel no desenvolvimento de DHGNA nas crianças obesas.
O equilíbrio entre os mecanismos pró‐inflamatórios e anti‐inflamatórios é de importância crítica para o desenvolvimento e a progressão da DHGNA. O estímulo de macrófagos pró‐inflamatórios por mediadores pró‐inflamatórios, tais como interferon‐γ e lipopolissacarídeos, estimula a secreção de citocinas pró‐inflamatórias, como TNFα, IL‐6, IL‐17 e IL‐23, o que por sua vez causa danos hepáticos e metabólicos.23,24 Em contraste, as células T regulatórias (Treg) impedem a produção de citocinas pró‐inflamatórias, como a IL‐17, ao expressar citocinas anti‐inflamatórias, como a IL‐10, o que controla a inflamação.25 Em estudos experimentais, a ativação da via da IL‐17 demonstrou que ela desempenha um papel significativo no desenvolvimento de DHGNA, na progressão da esteatose e na formação de fibrose.26
Sabe‐se que o polimorfismo IL‐17A rs2275913 é fortemente associado à secreção de IL‐17 pelas células‐T. No estudo in vitro de Espinoza et al., observou‐se um nível mais elevado de secreção de IL‐17 em células‐T estimuladas de indivíduos com o alelo AA de IL‐17A rs2275913 do que nos indivíduos sem o alelo.27 A ligação diferencial das variantes alélicas do polimorfismo IL‐17A rs2275913 ao fator nuclear de células T ativadas (NFAT) tem sido proposta como responsável pelas diferenças na secreção de IL‐17A.27 Várias doenças inflamatórias têm sido associadas clinicamente ao polimorfismo IL‐17A rs2275913. Observou‐se que o homozigoto rs2275913 AA apresenta risco aumentado de suscetibilidade à artrite reumatoide em populações caucasianas e colite ulcerativa em populações japonesas.28,29
No presente estudo, descobrimos que o polimorfismo IL‐17A rs2275913 foi mais frequente em crianças obesas com DHGNA do que naquelas sem DHGNA. Um estudo anterior também relatou uma associação entre a concentração de IL‐17 e a concentração de ALT em pacientes com infecção crônica pelo vírus da hepatite B, refletiu o grau de dano hepático.30 Consistentemente com essas observações, as concentrações séricas de ALT e AST diferiram significativamente entre os grupos de alelos de IL‐17 rs2275913 (AA, AG e GG). As concentrações séricas de ALT e AST foram mais altas no genótipo AA e mais baixas no genótipo GG. Esses achados sugerem que o genótipo AA pode ser um fator de risco para o desenvolvimento de DHGNA em pacientes obesos.
Não encontramos diferenças significativas nos polimorfismos dos genes MCP‐1 rs1024611, CCR‐2 rs1799864 e ABCA1 rs4149313 entre crianças obesas com e sem DHGNA, isso indicou que esses polimorfismos não parecem desempenhar um papel no desenvolvimento da doença. No entanto, esses polimorfismos podem estar relacionados ao desenvolvimento de esteato‐hepatite em crianças obesas, uma vez que não pudemos avaliar o grau de fibrose e inflamação hepática em nossos pacientes.
Este estudo tem algumas limitações. Primeiro, a população do estudo era relativamente pequena. Sabe‐se que a frequência dos polimorfismos estudados varia entre as etnias. A estrutura étnica da população poderia afetar a prevalência dos polimorfismos. Portanto, não podemos generalizar nossos resultados para outras populações. Em segundo lugar, a DHGNA foi diagnosticada por ultrassonografia e pelos níveis elevados de transaminases, que são consideradas ferramentas imperfeitas para detectar esteatose. Nenhum dos pacientes em nosso estudo foi submetido a biópsia hepática ou exame com FibroScan®. Portanto, nem a fibrose nem a inflamação do fígado puderam ser avaliadas nos participantes. Consequentemente, a associação entre os polimorfismos e o grau de fibrose e inflamação não pôde ser avaliada. Terceiro, o efeito do polimorfismo IL‐17A rs2275913 na transcrição gênica não foi investigado.
Em conclusão, os resultados deste estudo mostraram que o genótipo AA do gene IL‐17A pode estar associado ao desenvolvimento da DHGNA. Esse polimorfismo pode servir como um preditor para a esteatose hepática e fornecer informações úteis para identificar potenciais alvos terapêuticos para o tratamento de doenças hepáticas em pacientes obesos. Diferentemente do IL‐17A (‐197 G / A) (rs2275913), nenhuma associação foi encontrada entre os polimorfismos MCP‐1 (‐2518 A/G) (rs1024611), CCR‐2 (190 G/A) (rs1799864) e ABCA1 (883 G/A) (rs4149313) e DHGNA em crianças turcas obesas. Outros estudos de grande escala precisam ser feitos sobre a associação entre esses polimorfismos e a DHGNA, bem como a fibrose, em diferentes etnias, a fim de confirmar nossos achados.
FinanciamentoEste estudo foi financiado pelo Kanuni Training and Research Hospital.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Aprovação do comitê de éticaA aprovação do comitê de ética do Kanuni Training and Research Hospital foi recebida para este estudo.
Como citar este artigo: Akbulut UE, Emeksiz HC, Citli S, Cebi AH, Korkmaz HA, Baki G. IL‐17A, MCP‐1, CCR‐2, and ABCA1 polymorphisms in children with non‐alcoholic fatty liver disease. J Pediatr (Rio J). 2019;95:350–7.




