



To describe the results obtained in a neonatal screening program after its implementation and to assess the clinical and molecular profiles of confirmed and suspicious congenital adrenal hyperplasia cases.
MethodsA cross-sectional study was conducted. Newborns with suspected disease due to high 17-hydroxyprogesterone levels and adjusted for birth weight were selected. Classical congenital adrenal hyperplasia (salt-wasting and simple virilizing forms) was diagnosed by an increase in 17-hydroxyprogesterone levels as confirmed in the retest, clinical evaluation, and genotype determined by SNaPshot and multiplex ligation-dependent probe amplification.
ResultsAfter 24 months, 15 classic congenital adrenal hyperplasia cases were diagnosed in a total of 217,965 newborns, with an estimated incidence of 1:14,531. From 132 patients, seven non-classical and 14 heterozygous patients were screened for CYP21A2 mutations, and 96 patients presented false positives with wild type CYP21A2. On retest, increased 17-hydroxyprogesterone levels were found in classical congenital adrenal hyperplasia patients and showed significant correlation with genotype-related classical genital adrenal hyperplasia. The most frequent mutations were IVS2-13A/C>G followed by gene deletion or rearrangement events in the classical form. In non-classical and heterozygous diseases, p.Val282Leu was the most common mutation.
ConclusionsThe results underscore the effectiveness of congenital adrenal hyperplasia neonatal screening in the public health system and indicate that the adopted strategy was appropriate. The second sample collection along with genotyping of suspected cases helped to properly diagnose both severe and milder cases and delineate them from false positive patients.
Descrever os resultados obtidos em um programa de triagem neonatal após sua implementação e avaliar os perfis clínicos e moleculares de casos confirmados e suspeitos de hiperplasia adrenal congênita.
MétodosFoi feito um estudo transversal. Recém-nascidos com suspeita da doença devido aos altos níveis de 17-alfa-hidroxiprogesterona e ajustados pelo peso ao nascer foram selecionados. A hiperplasia adrenal congênita clássica (forma perdedora de sal e forma virilizante simples) foi diagnosticada por um aumento nos níveis de 17-alfa-hidroxiprogesterona confirmado no reteste, avaliação clínica e genótipo determinado com o uso do ensaio SNaPshot e amplificação multiplex de sondas dependente de ligação.
ResultadosApós 24 meses, 15 casos clássicos de hiperplasia adrenal congênita foram diagnosticados em 217.965 recém-nascidos, com uma incidência estimada de 1:14.531. De 132 pacientes, sete não clássicos e 14 heterozigotos foram submetidos à triagem para mutações no gene CYP21A2 e 96 pacientes apresentaram resultados falso‐positivos com CYP21A2 do tipo selvagem. No reteste, níveis aumentados de 17-alfa-hidroxiprogesterona foram encontrados em pacientes com hiperplasia adrenal congênita clássica e mostraram correlação significativa com HAC clássica relacionada ao genótipo. As mutações mais frequentes foram IVS2-13A/C>G, seguidas de deleção gênica ou eventos de rearranjo na forma clássica. Em casos de doenças não clássicas e heterozigose, a mutação p.Val282Leu foi a mais comum.
ConclusõesOs resultados ressaltam a eficácia da triagem neonatal para a hiperplasia adrenal congênita no sistema público de saúde e indicam que a estratégia adotada foi adequada. A segunda coleta de amostras, juntamente com a genotipagem dos casos suspeitos, ajudou a diagnosticar adequadamente os casos graves e mais leves e diferenciá-los de pacientes com resultado falso-positivo.
A hiperplasia adrenal congênita (HAC), doença autossômica recessiva, é caracterizada pelo comprometimento da síntese metabólica de cortisol e aldosterona.1 Ela é causada por mutações no gene CYP21A2 em aproximadamente 90% de todos os casos, leva à deficiência de 21‐hidroxilase (21‐OH) e causa acúmulo de precursores de androgênio.2 O principal marcador da doença é a 17‐alfa‐hidroxiprogesterona (17‐OHP).2 Existem três formas clínicas reconhecidas de HAC: forma clássica perdedora de sal (PS); virilizante simples (VS); e a forma tardia, denominada HAC não clássica (NC).1,3 O principal objetivo da triagem neonatal é identificar recém‐nascidos em risco de serem acometidos pelas duas formas clássicas.4,5 No entanto, casos assintomáticos com níveis persistentemente elevados de 17‐OHP na triagem neonatal são suspeitos para HAC‐NC e podem ser identificados por diagnóstico molecular.6,7
A incidência global de HAC descrita na literatura é de 1:15.000 em recém‐nascidos vivos.4 A prevalência de HAC‐NC é estimada em 1:1.000 indivíduos.1 Em 2/3 dos casos das formas clássicas, a perda de sal afeta ambos os gêneros e os sintomas começam na segunda semana de vida, reforça a importância do diagnóstico precoce.5 Nas meninas, a exposição excessiva ao androgênio intrauterino causa diferentes graus de virilização e ambiguidade genital, conforme definido pela Escala de Prader.8 Aumentos na 17‐OHP foram observados em recém‐nascidos sem HAC (falso‐positivos) devido a situações estressantes e parto prematuro.9,10 Por sua vez, casos falsos negativos podem ocorrer como resultado do uso materno de corticosteroides no fim da gravidez.11,12 Para minimizar o número de falso‐positivos, foram estabelecidos pontos de corte estratificados por categorias brasileiras e internacionais de peso ao nascer.9–11
A análise molecular do gene CYP21A2 pode melhorar a especificidade da triagem neonatal e auxiliar no manejo clínico da doença.7,13 As principais mutações do gene CYP21A2 foram categorizadas em quatro grupos (Null, A, B e C), de acordo com a atividade enzimática residual da 21‐OH, o que permite a determinação do fenótipo esperado.2,6,14,15 Tanto o tipo Null quanto o A estão associados ao HAC‐PS, o grupo B ao HAC‐VS e o grupo C ao HAC‐NC.2,6,13–15
No Brasil, Goiás, Santa Catarina e São Paulo acumularam mais de 10 anos de experiência em triagem de HAC neonatal.9,14,16,17 Assim, os objetivos deste estudo foram: 1) descrever os resultados 24 meses após a triagem neonatal de HAC em um programa de saúde pública no Sul do Brasil; e 2) avaliar os quadros clínicos de casos confirmados e suspeitos de HAC, além da genotipagem molecular desses casos como uma ferramenta complementar para melhorar o diagnóstico da HAC.
MétodosFoi feito um estudo transversal com recém‐nascidos incluídos nos dois primeiros anos da implantação do programa público de triagem de HAC no Estado do Rio Grande do Sul, Brasil. Foi revisado um banco de dados de bebês com suspeita de HAC, baseado em valores alterados de 17‐OHP a partir de uma triagem feita de 2014 a 2016. A HAC clássica (HAC‐C), PS e VS, foi diagnosticada pelo aumento dos níveis de 17‐OHP detectados na triagem, confirmado por reteste e/ou avaliação clínica, seguido por estudos genotípicos. As meninas afetadas apresentaram genitália externa virilizada, enquanto os meninos não necessariamente apresentaram alterações genitais, mas o aumento do pênis pôde ser observado. Os falsos positivos foram caracterizados por níveis mais baixos de 17‐OHP no reteste e pelo genótipo do tipo selvagem (WT, do inglês wild type), de acordo com a figura 1.18 Os dados relacionados à distribuição dos genótipos foram relatados recentemente.19

Fluxograma da avaliação genótipo‐fenótipo da HAC. 17 OHP, 17‐alfa‐hidroxiprogesterona; HAC, hiperplasia adrenal congênita. Adaptado de Kopacek et al.18
O protocolo do estudo foi aprovado pelo Comitê de Ética em Pesquisa do Hospital Materno‐Infantil Presidente Vargas e pelo Comitê de Ética em Pesquisa da Fundação Estadual de Produção e Pesquisa em Saúde (n° 341.289/8 de junho de 2013); todos os pais assinaram o termo de consentimento livre e esclarecido.
Amostras de sangue seco foram obtidas com papel de filtro (S&S 903) do 3° ao 40° dia do pós‐natal. O ensaio de imunofluorescência GSP de fase sólida (time‐resolved) (kit Neonatal 17‐OHP; Perkin‐Elmer, Turku, Finlândia) foi usado para medir a 17‐OHP. A figura 1 descreve a estratégia do estudo.9,11,18 Os recém‐nascidos submetidos ao uso de corticosteroides maternos no fim da gestação, conforme registrado em papel de filtro, foram convocados para uma segunda coleta após 15 dias de vida.11,12,18
A genotipagem foi feita conforme descrito anteriormente por Prado et al.19 em 132 indivíduos. O ensaio SNaPshot foi usado para todos os indivíduos. Esse ensaio analisou 12 mutações de ponto no gene CYP21A2 (p.Gln319Ter, p.Arg357Trp, p.Leu308Phefs, p.Val237Glu, IVS2‐13A/C>G, p.Ile173Asn, p.Pro31Leu, p.Pro454Ser, p.Val282Leu, p.Gly111Valfs, p.Arg409Cys e p.His63Leu).19–21 Para casos com suspeita clínica e/ou bioquímica de HAC clássica, mas nenhuma mutação identificada pela técnica anterior, um kit comercial de amplificação multiplex de sondas dependente de ligação (MLPA, do inglês Multiplex Ligation‐dependent Probe Amplification) com o kit SALSA MLPA probemix P050‐C1 CAH (MRC Holland, Amsterdã, Holanda) foi usado para detectar eventos de rearranjos.
Os resultados foram expressos como média + desvio padrão (DP) ou frequências (%). As comparações entre os grupos foram analisadas por análise de variância (Anova) univariada seguida pelo teste de Tukey. A análise de covariância (Ancova) foi usada para ajustes de 17‐OHP por peso ao nascer e os dados foram expressos como média + erro padrão da média (EPM). Uma correlação parcial foi estimada entre 17‐OHP e eletrólitos. As análises foram feitas com o programa SPSS (PASW Estatística para Windows, Versão 18.0. Chicago, EUA). Os dados foram considerados significativos quando p < 0,05.
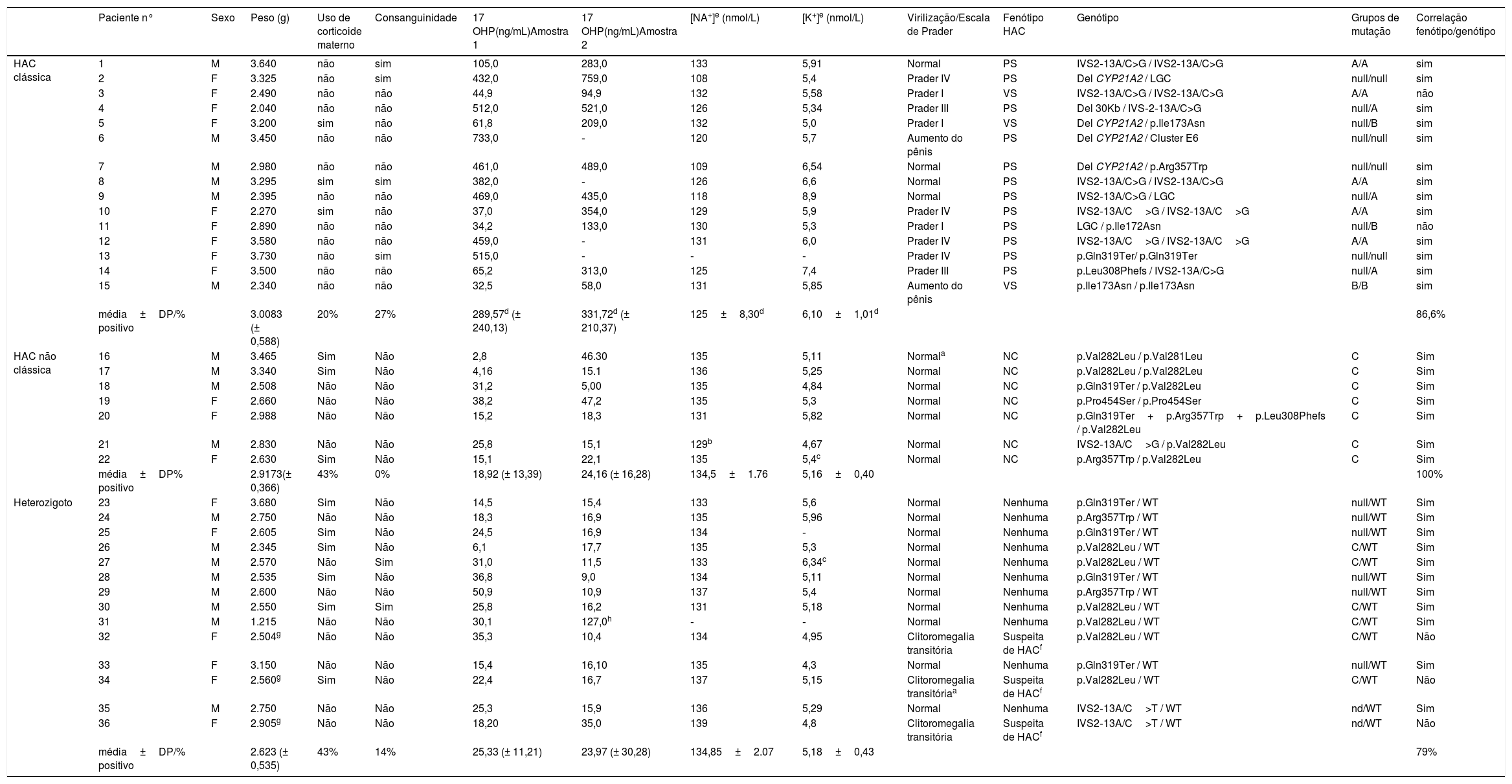
ResultadosDe 217.965 amostras obtidas na triagem de dois anos, 15 casos de HAC‐C foram diagnosticados, com uma incidência estimada de 1:14.531. Nove recém‐nascidos eram do sexo feminino e seis do masculino e 80% (n = 12) foram clinicamente caracterizados como pacientes com HAC‐PS e o restante como pacientes com a forma VS. De acordo com nossos dados publicados anteriormente,18 70‐80% da primeira amostra tinham sido coletados entre três e sete dias de vida e esse achado foi semelhante ao HAC‐C [mediana 5 (2,0 a 38,0) dias]. A maioria das amostras de reteste foi coletada em torno da segunda ou terceira semana de vida [mediana 17 (14,0‐21,0) dias], semelhantemente à HAC‐C [mediana 19 (8,0–51,0) dias]. A tabela 1 descreve os dados clínicos, os valores de 17‐OHP e eletrólitos e a avaliação fenotípica‐genotípica desses casos. Três meninas (casos 3, 5 e 11) apresentaram virilização leve (classificação de Prader I) e não foram caracterizadas clinicamente como portadoras de clitoromegalia antes da triagem da HAC. Outra menina (caso 2) foi designada como um menino e a outra (caso 10) foi registrada com um nome neutro, adequado para os dois sexos, devido à ambiguidade genital. Nas outras quatro meninas (casos 4, 12, 13 e 14), a ambiguidade genital foi identificada, mas a HAC não foi diagnosticada antes da triagem. Entre os meninos, os casos 6 e 15 apresentaram aumento do pênis e os outros quatro tinham genitália masculina normal. Dez dos 15 casos (66,6%) nasceram com peso > 2.500g e nenhum caso pesava < 2.000g. As mães de três casos (5, 8 e 10) usavam glicocorticoides no fim de suas gestações. Em dois casos, foi observado um aumento líquido nos níveis de 17‐OHP na segunda amostra. Em um terceiro lactente (caso 8), o início da corticoterapia por perda de sal antecedeu a possibilidade de obtenção de uma segunda amostra. Quatro bebês (26,6%) tinham pais consanguíneos.
Aspectos clínicos e genótipos em pacientes com HAC clássica, não clássica e heterozigótica
| Paciente n° | Sexo | Peso (g) | Uso de corticoide materno | Consanguinidade | 17 OHP(ng/mL)Amostra 1 | 17 OHP(ng/mL)Amostra 2 | [NA+]e (nmol/L) | [K+]e (nmol/L) | Virilização/Escala de Prader | Fenótipo HAC | Genótipo | Grupos de mutação | Correlação fenótipo/genótipo | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HAC clássica | 1 | M | 3.640 | não | sim | 105,0 | 283,0 | 133 | 5,91 | Normal | PS | IVS2‐13A/C>G / IVS2‐13A/C>G | A/A | sim |
| 2 | F | 3.325 | não | sim | 432,0 | 759,0 | 108 | 5,4 | Prader IV | PS | Del CYP21A2 / LGC | null/null | sim | |
| 3 | F | 2.490 | não | não | 44,9 | 94,9 | 132 | 5,58 | Prader I | VS | IVS2‐13A/C>G / IVS2‐13A/C>G | A/A | não | |
| 4 | F | 2.040 | não | não | 512,0 | 521,0 | 126 | 5,34 | Prader III | PS | Del 30Kb / IVS‐2‐13A/C>G | null/A | sim | |
| 5 | F | 3.200 | sim | não | 61,8 | 209,0 | 132 | 5,0 | Prader I | VS | Del CYP21A2 / p.Ile173Asn | null/B | sim | |
| 6 | M | 3.450 | não | não | 733,0 | ‐ | 120 | 5,7 | Aumento do pênis | PS | Del CYP21A2 / Cluster E6 | null/null | sim | |
| 7 | M | 2.980 | não | não | 461,0 | 489,0 | 109 | 6,54 | Normal | PS | Del CYP21A2 / p.Arg357Trp | null/null | sim | |
| 8 | M | 3.295 | sim | sim | 382,0 | ‐ | 126 | 6,6 | Normal | PS | IVS2‐13A/C>G / IVS2‐13A/C>G | A/A | sim | |
| 9 | M | 2.395 | não | não | 469,0 | 435,0 | 118 | 8,9 | Normal | PS | IVS2‐13A/C>G / LGC | null/A | sim | |
| 10 | F | 2.270 | sim | não | 37,0 | 354,0 | 129 | 5,9 | Prader IV | PS | IVS2‐13A/C>G / IVS2‐13A/C>G | A/A | sim | |
| 11 | F | 2.890 | não | não | 34,2 | 133,0 | 130 | 5,3 | Prader I | PS | LGC / p.Ile172Asn | null/B | não | |
| 12 | F | 3.580 | não | não | 459,0 | ‐ | 131 | 6,0 | Prader IV | PS | IVS2‐13A/C>G / IVS2‐13A/C>G | A/A | sim | |
| 13 | F | 3.730 | não | sim | 515,0 | ‐ | ‐ | ‐ | Prader IV | PS | p.Gln319Ter/ p.Gln319Ter | null/null | sim | |
| 14 | F | 3.500 | não | não | 65,2 | 313,0 | 125 | 7,4 | Prader III | PS | p.Leu308Phefs / IVS2‐13A/C>G | null/A | sim | |
| 15 | M | 2.340 | não | não | 32,5 | 58,0 | 131 | 5,85 | Aumento do pênis | VS | p.Ile173Asn / p.Ile173Asn | B/B | sim | |
| média±DP/% positivo | 3.0083 (± 0,588) | 20% | 27% | 289,57d (± 240,13) | 331,72d (± 210,37) | 125±8,30d | 6,10±1,01d | 86,6% | ||||||
| HAC não clássica | 16 | M | 3.465 | Sim | Não | 2,8 | 46.30 | 135 | 5,11 | Normala | NC | p.Val282Leu / p.Val281Leu | C | Sim |
| 17 | M | 3.340 | Sim | Não | 4,16 | 15.1 | 136 | 5,25 | Normal | NC | p.Val282Leu / p.Val282Leu | C | Sim | |
| 18 | M | 2.508 | Não | Não | 31,2 | 5,00 | 135 | 4,84 | Normal | NC | p.Gln319Ter / p.Val282Leu | C | Sim | |
| 19 | F | 2.660 | Não | Não | 38,2 | 47,2 | 135 | 5,3 | Normal | NC | p.Pro454Ser / p.Pro454Ser | C | Sim | |
| 20 | F | 2.988 | Não | Não | 15,2 | 18,3 | 131 | 5,82 | Normal | NC | p.Gln319Ter+p.Arg357Trp+p.Leu308Phefs / p.Val282Leu | C | Sim | |
| 21 | M | 2.830 | Não | Não | 25,8 | 15,1 | 129b | 4,67 | Normal | NC | IVS2‐13A/C>G / p.Val282Leu | C | Sim | |
| 22 | F | 2.630 | Sim | Não | 15,1 | 22,1 | 135 | 5,4c | Normal | NC | p.Arg357Trp / p.Val282Leu | C | Sim | |
| média±DP% positivo | 2.9173(± 0,366) | 43% | 0% | 18,92 (± 13,39) | 24,16 (± 16,28) | 134,5±1.76 | 5,16±0,40 | 100% | ||||||
| Heterozigoto | 23 | F | 3.680 | Sim | Não | 14,5 | 15,4 | 133 | 5,6 | Normal | Nenhuma | p.Gln319Ter / WT | null/WT | Sim |
| 24 | M | 2.750 | Não | Não | 18,3 | 16,9 | 135 | 5,96 | Normal | Nenhuma | p.Arg357Trp / WT | null/WT | Sim | |
| 25 | F | 2.605 | Sim | Não | 24,5 | 16,9 | 134 | ‐ | Normal | Nenhuma | p.Gln319Ter / WT | null/WT | Sim | |
| 26 | M | 2.345 | Sim | Não | 6,1 | 17,7 | 135 | 5,3 | Normal | Nenhuma | p.Val282Leu / WT | C/WT | Sim | |
| 27 | M | 2.570 | Não | Sim | 31,0 | 11,5 | 133 | 6,34c | Normal | Nenhuma | p.Val282Leu / WT | C/WT | Sim | |
| 28 | M | 2.535 | Sim | Não | 36,8 | 9,0 | 134 | 5,11 | Normal | Nenhuma | p.Gln319Ter / WT | null/WT | Sim | |
| 29 | M | 2.600 | Não | Não | 50,9 | 10,9 | 137 | 5,4 | Normal | Nenhuma | p.Arg357Trp / WT | null/WT | Sim | |
| 30 | M | 2.550 | Sim | Sim | 25,8 | 16,2 | 131 | 5,18 | Normal | Nenhuma | p.Val282Leu / WT | C/WT | Sim | |
| 31 | M | 1.215 | Não | Não | 30,1 | 127,0h | ‐ | ‐ | Normal | Nenhuma | p.Val282Leu / WT | C/WT | Sim | |
| 32 | F | 2.504g | Não | Não | 35,3 | 10,4 | 134 | 4,95 | Clitoromegalia transitória | Suspeita de HACf | p.Val282Leu / WT | C/WT | Não | |
| 33 | F | 3.150 | Não | Não | 15,4 | 16,10 | 135 | 4,3 | Normal | Nenhuma | p.Gln319Ter / WT | null/WT | Sim | |
| 34 | F | 2.560g | Sim | Não | 22,4 | 16,7 | 137 | 5,15 | Clitoromegalia transitóriaa | Suspeita de HACf | p.Val282Leu / WT | C/WT | Não | |
| 35 | M | 2.750 | Não | Não | 25,3 | 15,9 | 136 | 5,29 | Normal | Nenhuma | IVS2‐13A/C>T / WT | nd/WT | Sim | |
| 36 | F | 2.905g | Não | Não | 18,20 | 35,0 | 139 | 4,8 | Clitoromegalia transitória | Suspeita de HACf | IVS2‐13A/C>T / WT | nd/WT | Não | |
| média±DP/% positivo | 2.623 (± 0,535) | 43% | 14% | 25,33 (± 11,21) | 23,97 (± 30,28) | 134,85±2.07 | 5,18±0,43 | 79% |
F, sexo feminino; HAC, hiperplasia adrenal congênita; HT, heterozigoto; LGC, Large Gene Conversion; M, sexo masculino; NC, não clássica; nd, não definido.
A tabela 1 também descreve o perfil clínico, os valores de 17‐OHP e a avaliação do fenótipo‐genótipo dos casos de HAC‐NC e heterozigotos que foram detectados durante a triagem. Apenas um paciente heterozigoto (caso 31) apresentou baixo peso ao nascer relacionado à prematuridade extrema e valor aumentado de 17‐OHP na segunda amostra, associado à pioria clínica na unidade intensiva neonatal. Esse paciente foi acompanhado em um hospital terciário distante do centro de triagem. Foram relatados valores eletrolíticos normais sem qualquer outro sinal clínico de HAC por esse hospital. Uma terceira amostra foi coletada após melhoria clínica e mostrou uma diminuição considerável de 17‐OHP (17 ng/mL). Nesse paciente, o genótipo foi crucial para a elucidação do diagnóstico. Outros três pacientes com HAC‐NC (16, 17 e 22) apresentaram valores maiores de 17‐OHP na segunda amostra e um histórico de uso de corticosteroide pela mãe. Não foram observadas hiponatremia e hipercalemia nos casos de HAC‐NC e heterozigotos. Entre os casos iniciais suspeitos de HAC, um paciente com hiperpigmentação genital transitória (paciente 16) foi diagnosticado como HAC‐NC e três meninas com clitoromegalia transitória foram posteriormente identificadas como casos heterozigotos. Nesses casos, a regressão espontânea da clitoromegalia foi observada durante o seguimento até os primeiros seis meses de vida. A consanguinidade dos pais foi encontrada em dois (14,3%) dos pacientes heterozigotos.
A tabela 1 mostra a distribuição dos genótipos de acordo com os grupos de atividade enzimática (Null, A, B e C) e heterozigosidade na população de recém‐nascidos. A concordância genótipo‐fenótipo foi observada em 13 casos de HAC‐C (86,6%). A mutação IVS2‐13A/C>G foi a mais frequente (33% em homozigose e 20% em heterozigosidade composta), seguida pela p.Ile173Asn, deleção e conversão gênica, cada uma observada em 20% dos casos de HAC‐C. Sete pacientes apresentaram mutações não clássicas (grupo C) e os outros 14 pacientes heterozigotos tiveram o alelo WT detectado durante a triagem neonatal. Na amostra total, a mutação p.Val282Leu foi a mais frequente nos pacientes com HAC‐NC (57% heterozigotos e 28,5% homozigotos) e também em seis pacientes heterozigotos (43%) (tabela 1). Um novo alelo IVS2‐13A/C>T foi descrito por Prado et al. em uma menina heterozigota nascida a termo com peso ao nascer de 2.905g (amostra 1: 17 OHP 18,2 ng/mL e amostra 2: 35,0 ng/mL) e em um menino prematuro tardio com peso de nascimento de 2.540g (amostra 1: 17‐OHP 25,3 ng/mL e amostra 2: 15,9 ng/mL) com triagem positiva.19 Surpreendentemente, a clitoromegalia identificada na menina durante o primeiro exame foi resolvida espontaneamente durante o período de seguimento. Ambos apresentaram perfil eletrolítico normal e não tinham quaisquer outros sintomas associados à HAC durante o seguimento de seis meses.
Os níveis de 17‐OHP, ajustados para o peso ao nascer, foram analisados de acordo com os grupos (1 = HAC‐C; 2 = HAC‐NC; 3 = heterozigoto; 4 = WT) nas amostras 1 e 2 (fig. 2A e B, respectivamente). Foram observados níveis significativamente mais altos de 17‐OHP na HAC‐C em comparação com outros grupos nas amostras 1 e 2. A figura 3 mostra os níveis de 17‐OHP, ajustados para o peso ao nascer, nas amostras 1 e 2, classificados de acordo com os grupos de mutação. Na amostra 1, indivíduos com grupos de mutação Null e A apresentaram níveis significativamente mais altos de 17‐OHP e na amostra 2, os grupos de mutação Null, A e B mantiveram níveis mais altos de 17‐OHP, os quais foram estatisticamente diferentes do grupo C (HAC‐NC), heterozigotos e pacientes WT.
. Quatro grupos (1 = HAC clássica; 2 = HAC NC; 3 = heterozigoto; 4 = Tipo selvagem). A, Níveis de 17‐OHP na amostra 1; B, Níveis de 17‐OHP na amostra 2. HAC, hiperplasia adrenal congênita; NC, não clássica. a p < 0,05 versus todos os outros grupos.")
Níveis de 17‐OHP nos quatro grupos (HAC clássica, HAC‐NC, heterozigoto e tipo selvagem). Quatro grupos (1 = HAC clássica; 2 = HAC NC; 3 = heterozigoto; 4 = Tipo selvagem). A, Níveis de 17‐OHP na amostra 1; B, Níveis de 17‐OHP na amostra 2. HAC, hiperplasia adrenal congênita; NC, não clássica. a p < 0,05 versus todos os outros grupos.
; A. Níveis de 17‐OHP na amostra 1; B. Níveis de 17‐OHP na amostra 2. Genótipo (1 = Grupo Null; 2 = Grupo A; 3 = Grupo B; 4 = Grupo C; 5 = Heterozigoto; 6 = Tipo Selvagem). a p < 0,05 versus todos os outros grupos. b p < 0,05 versus grupo 1 e 2. HAC, hiperplasia adrenal congênita; 17‐OHP, 17‐alfa‐hidroxiprogesterona; PS, perdedora de sal; VS, virilizante simples; HAC‐C, hiperplasia adrenal congênita clássica; NC, não clássica, de início tardio; WT, tipo selvagem; SPSS, Statistical Package for the Social Sciences.")
Níveis de 17‐OHP de acordo com o genótipo (grupo de gravidade de mutação, heterozigose e tipo selvagem); A. Níveis de 17‐OHP na amostra 1; B. Níveis de 17‐OHP na amostra 2. Genótipo (1 = Grupo Null; 2 = Grupo A; 3 = Grupo B; 4 = Grupo C; 5 = Heterozigoto; 6 = Tipo Selvagem). a p < 0,05 versus todos os outros grupos. b p < 0,05 versus grupo 1 e 2. HAC, hiperplasia adrenal congênita; 17‐OHP, 17‐alfa‐hidroxiprogesterona; PS, perdedora de sal; VS, virilizante simples; HAC‐C, hiperplasia adrenal congênita clássica; NC, não clássica, de início tardio; WT, tipo selvagem; SPSS, Statistical Package for the Social Sciences.
A tabela 1 também mostra os valores eletrolíticos nos diferentes grupos. Os casos de HAC‐C apresentaram níveis mais elevados de Na+ e K+. Foi encontrada uma forte correlação inversa entre o 17‐OHP ajustado para o peso ao nascer e o Na+ (r = −0,795 para Amostra 1 de 17‐OHP e r = −0,740, para Amostra 2, p < 0,05) e uma correlação positiva com K+ (r = 0,494 para Amostra 1 de 17‐OHP e r = 0,531 para Amostra 2, p <0,05).
DiscussãoNo presente estudo, o programa de triagem neonatal de dois anos da HAC que usou níveis significativamente mais altos do marcador, a 17‐OHP, permitiu de maneira adequada o diagnóstico de 15 recém‐nascidos com HAC clássica. Além disso, o reteste foi bem discriminado entre os casos e a gravidade da doença apresentou concordância com a investigação do genótipo correspondente.
O programa de triagem foi altamente eficaz na detecção de alguns casos que não seriam clinicamente reconhecidos antes da triagem. Nesse sentido, atipias genitais leves (casos 3, 4 e 11) e aumento do tamanho do pênis em meninos (casos 6 e 15) não foram detectados antes da avaliação especializada. Mesmo naqueles com genitália ambígua, a HAC não foi diagnosticada antes da triagem, a designação sexual incorreta foi feita em dois casos (2 e 10). De fato, observamos que mais de 50% das meninas tiveram uma avaliação clínica incorreta das características de virilização, o que foi identificado de maneira similar em estudos anteriores.9,15,22 Isso reforça a importância da triagem neonatal universal para hiperplasia no Brasil.
Em relação à grande diferença observada nos valores de corte de 17‐OHP usados para as categorias de peso ao nascer,9,11 é importante observar que os lactentes das categorias de menor peso são comumente internados em unidades de terapia intensiva e continuamente monitorados, inclusive para distúrbios eletrolíticos. De fato, como observado anteriormente durante o primeiro ano de triagem, a maioria dos falso‐positivos era de recém‐nascidos com peso < 2.000g.18 Uma segunda amostra que mostrou níveis mais baixos de 17‐OHP em casos assintomáticos é geralmente suficiente para elucidar os casos falso‐positivos, especialmente em bebês prematuros e recém‐nascidos de baixo peso. Além disso, como relatado anteriormente, as taxas de consanguinidade são maiores entre os casos de HAC.18,23 Dessa forma, essa informação adicional pode ser útil para distinguir os casos reais dos casos falso‐positivos. Da mesma forma, informações sobre o uso de corticoide pela mãe no fim da gestação são relevantes. Nesse caso, a coleta de uma segunda amostra após 15 dias de vida do recém‐nascido ajuda a diagnosticar casos como o do paciente 11, que apresentou virilização leve não reconhecida anteriormente e posterior desenvolvimento da HAC‐PS.22
Altos níveis de 17‐OHP podem estar presentes em recém‐nascidos prematuros e criticamente doentes, o que reforça a triagem estratificada pelo peso ao nascer.9–11 Além disso, o genótipo foi recomendado para melhorar o diagnóstico da precisão da triagem neonatal. Ele ajuda a diferenciar os casos reais de falso‐positivos, esclarece casos limítrofes e, eventualmente, permite que portadores de HAC‐NC sejam diagnosticados.7,13,15 Nos casos confirmados, valores elevados de 17‐OHP corresponderam à gravidade de cada grupo genotípico.14,15,22 Curiosamente, quando os níveis de 17‐OHP foram estratificados de acordo com a gravidade do grupo de mutações na segunda amostra, foi observada uma melhor diferenciação das formas VS (mutações do grupo B) da HAC‐NC (mutações do grupo C), heterozigotos e WT (figs. 2 e 3). Esses três últimos grupos apresentam níveis de 17‐OHP significativamente menores, o que justifica a estratégia de retestar esses pacientes e, quando ainda houver dúvidas, fazer a análise do genótipo.22
A eficácia do programa de triagem também pode ser demonstrada quando formas graves, como portadores de mutações do grupo Null, são identificadas.15 Deleção e grandes conversões gênicas que caracterizaram o grupo Null foram observadas em 20% da nossa amostra, o que está de acordo com um recente e grande estudo de correlação genótipo‐fenótipo.24 O grupo de mutações Null em homozigose foi observado em 26,67% de nossa amostra (4/15 pacientes) e nos grupos Null e A em heterozigose composta, o que caracterizou a HAC‐PS, em outros três pacientes. No paciente 11, não houve correlações fenótipo‐genótipo claras como resultado da manifestação de um fenótipo de HAC‐PS leve. Quando as mutações dos grupos Null e B estão em heterozigose composta, inclusive a p.Ile173Asn em um alelo, a HAC‐VS é esperada na maioria dos casos.6,15 Essa observação foi relatada por outros grupos,24,25 inclusive uma associação muito rara da mutação p.Ile173Asn com uma HAC‐NC.24 A HAC‐PS associada à p.Ile173Asn é mais comum quando uma segunda mutação mais grave está associada em heterozigosidade.25,26
No Brasil, um estudo de Bachega et al. determinou a frequência de mutações de ponto em 130 pacientes com HAC‐C e HAC‐NC e estabeleceu uma correlação genótipo‐fenótipo.27 As mutações mais frequentes foram a IVS2‐13A/C>G, em 55% dos alelos dos pacientes com a forma PS, a p.Ile173Asn, em 42% das HAC‐VS, e a pVal282Leu, em 70% dos indivíduos com HAC‐NC. A frequência da mutação IVS2‐13A/C>G em nossa amostra não foi, portanto, surpreendente para a população brasileira (53% em homo e/ou heterozigose na HAC‐C). Em uma amostra brasileira de São Paulo14 e em um estudo de coorte maior na Argentina, a IVS2‐13A/C> G correspondeu a 21% na frequência alélica e a 20% das mutações em homozigose,28 respectivamente. O Brasil e a Argentina (AR) apresentam taxas muito semelhantes dessa mutação.14,28 O Estado do Rio Grande do Sul está próximo da AR e a população compartilha origens étnicas comuns. Esse grupo também descreve a associação dos alelos IVS2‐13A/C>G com as formas VS,8 o que pode explicar o fenótipo‐genótipo não concordante no caso 32.
A nova variante IVS2‐13A/C>T foi identificada em dois pacientes. O alelo T foi observado nos casos em heterozigose com o alelo C benigno, como descrito anteriormente.19 Como a paciente do sexo feminino apresentou clitoromegalia ao nascimento, mesmo em casos heterozigotos, esse achado não pode ser negligenciado e requer estudos genéticos adicionais. A descrição dos três casos com clitoromegalia transitória em nossa amostra é digna de estudo adicional. Tem sido relatado que alguns pacientes heterozigotos apresentaram níveis mais elevados de andrógenos, especialmente os portadores da mutação pVal282Leu com adrenarca precoce.1,29 Espera‐se que os pacientes com HAC‐NC e pacientes heterozigotos sejam assintomáticos ao nascimento, mas nossa hipótese é que possíveis elevações androgênicas transitórias podem ocorrer. Estudos futuros com vistas a esclarecer esse fenômeno podem usar a genotipagem expandida pelo sequenciamento completo do gene CYP21A2 para excluir outras mutações não detectadas pelos ensaios SNaPshot e MLPA. Em nossa amostra, um melhor entendimento dessa nova variante pode fornecer informações mais específicas.
Um dos pontos fortes deste estudo foi que os diagnósticos e exames de acompanhamento foram feitos pelo mesmo endocrinologista pediátrico. Portanto, a avaliação clínica adequada dos pacientes feita neste estudo é apropriada para a caracterização clínica e molecular dessa amostra na triagem neonatal da HAC. Outro ponto forte foi o fato de os dados terem sido coletados a partir de um programa de triagem populacional, o que favoreceu a avaliação de um grande número de indivíduos. Nesse contexto, nosso estudo foi o primeiro programa de triagem neonatal/genotipagem com o uso da metodologia SNaPshot.19 Essa estratégia permite detectar simultaneamente as 12 mutações mais comumente associadas à HAC de forma rápida e ágil e agrega qualidade ao programa de triagem neonatal. Esse grupo de mutações de ponto representa 89% das mutações detectadas em nossa coorte do sul do Brasil.19
Em conclusão, os resultados deste estudo ressaltam a eficácia do programa de triagem na detecção de casos de HAC e na exclusão de casos suspeitos com base em pontos de corte dos níveis de 17‐OHP ligados à estratificação pelo peso ao nascer. Além disso, a coleta da segunda amostra, seguida pela genotipagem das amostras suspeitas, ajudou a diagnosticar adequadamente casos graves de HAC‐ PS/VS e casos mais leves de VS, além de diferenciar entre casos de HAC‐C e pacientes falsos positivos. Os presentes resultados também indicaram que a genotipagem é uma ferramenta diagnóstica valiosa e complementar para a triagem neonatal, a qual fornece informações sobre a gravidade da doença, permite o aconselhamento genético em casos graves e evita o tratamento excessivo da HAC‐NC de início tardio e dos pacientes falsos positivos.
FinanciamentoEste trabalho foi financiado por bolsas de estudo do Instituto Nacional Brasileiro de Hormônios e Saúde da Mulher, Conselho Nacional de Desenvolvimento Científico e Tecnológico [CNPq INCT 573747/2008‐3], do Sistema Nacional Brasileiro de Saúde Pública (PPSUS) e da Fundação de Amparo à Pesquisa do Rio Grande do Sul (Fapergs), Porto Alegre, Brasil.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Kopacek C, Prado MJ, da Silva CM, de Castro SM, Beltrão LA, Vargas PR, et al. Clinical and molecular profile of newborns with confirmed or suspicious congenital adrenal hyperplasia detected after a public screening program implementation. J Pediatr (Rio J). 2019;95:282–90.
Estudo feito no Serviço de Referência em Triagem Neonatal (centro de referência para triagem neonatal do Sistema Público de Saúde) do Hospital Materno Infantil Presidente Vargas e no Centro de Desenvolvimento Científico e Tecnológico (CDCT), Porto Alegre, RS, Brasil.




