





To verify and describe the main events related to the diagnosis and management of chronic obstructive pulmonary diseases in children (COPDC) and adolescents, considering the interrelated physiopathology, genetic, and environmental characteristics.
SourcesRelevant literature from PubMed was selected and reviewed.
Summary of the findingsCOPDC have an environmental and/or genetic origin and its manifestation has manifold genotypes, phenotypes, and endotypes. Although COPDC has no cure, it can be clinically controlled. Chronic cough is the main symptom and bronchiectasis can be present in several COPDC patients. The management of COPDC is more effective if based on guidelines and when treatment regimen adherence is promoted. Oral and inhaled corticosteroids, bronchodilators, inhaled antibiotics, and treatment of pulmonary exacerbation (PE) are the bases of COPDC management, and should be individualized for each patient.
ConclusionsCorrect diagnosis and knowledge of risk factors and comorbidities are essential in COPDC management. Procedures and drugs used should be based on specific guidelines for each COPDC case. Treatment adherence is critical to obtain the benefits of management. COPDC clinical control must be evaluated by the decrease in PEs, improved quality of life, reduction of pulmonary function loss, and lung structural damage. For most cases of COPDC, monitoring by interdisciplinary teams in specialized reference centers with surveillance strategies and continuous care leads to better outcomes, which must be evaluated by decreasing pulmonary function damage and deterioration, better prognosis, better quality life, and increased life expectancy.
Verificar e descrever os principais eventos relacionados ao diagnóstico e manejo das doenças pulmonares obstrutivas crônicas em crianças (DPOCC) e adolescentes, tendo em vista a fisiopatologia, características genéticas e ambientais inter-relacionadas.
Fonte dos dadosRevisão na base de dado PUBMED com seleção de referências relevantes.
Síntese dos dadosAs DPOCC têm origem ambiental e/ou genética e se manifestam com diversos genótipos, fenótipos e endótipos e, embora possam ser controladas, não têm cura. O principal sintoma é a tosse crônica e muitas cursam com bronquiectasia. O manejo tem maior eficácia se baseado em guidelines e se a adesão ao regime terapêutico for estimulada e comprovada. Corticóides orais e inalatórios, broncodilatadores, antibióticos inalados e tratamento das exacerbações pulmonares (EP) são vigas mestras do manejo e devem ser individualizados para cada DPOCC.
ConclusõesNas DPOCC é fundamental o diagnóstico correto, conhecer os fatores de risco e comorbidades. Os procedimentos e os medicamentos devem ser baseados em guidelines específicos para cada DPOCC. Adesão ao tratamento é fundamental para obter os benefícios do manejo. O controle deve ser avaliado pela diminuição das EP, melhora na qualidade de vida e redução da evolução da perda da função e dano estrutural pulmonar. Para a maioria das DPOCC, o acompanhamento por equipes interdisciplinares em centros de referência especializados, com estratégias de vigilância e acolhimento contínuos, conduz a melhores desfechos que devem ser avaliados pela diminuição da deterioração do dano e da função pulmonar, melhor prognóstico, melhor qualidade de vida e aumento da expectativa de vida.
Chronic obstructive pulmonary disease (COPD) is typically shown in the literature as evidence, in most cases, of the damage caused by smoking in adults older than 40 years. Without any direct and exclusive association with active long-term smoking, several chronic obstructive pulmonary diseases in children (COPDC) and adolescents progress with deterioration in lung structure and function, causing persistent (fixed) or intermittent (temporary) obstruction to pulmonary flow, secondary to genetic and/or environmental changes that cause airway inflammation and/or infection. Although the symptoms of COPDC are very similar, they have variable etiology, morbidity, physiopathology, prevalence, prognosis, genotypes, and phenotypes.1,2
While some COPDC have been the object of many studies, such as asthma, cystic fibrosis (CF), recurrent wheezing in infants (RWI), and bronchopulmonary dysplasia (BPD), others are known as “orphan diseases,” such as primary ciliary dyskinesia (PCD), non-cystic fibrosis bronchiectasis (NCFB), plastic bronchitis (PB), and bronchiolitis obliterans (BO).
COPDC are characterized by high prevalence of asthma, RWI, and BPD, or low prevalence of BO, CF, PB, PCD, and NCFB. They are noncommunicable diseases, of long duration and slow progression, showing episodes of pulmonary exacerbation (PE), acute or permanent airflow limitation, and significant quality of life impairment.3 In all, the main pulmonary symptom is chronic cough, reflecting the presence of alterations in the airways, as there are no cough receptors in the alveoli. Another characteristic is the presence of bronchiectasis in many of them.4
Most of them, including some of the several clinical forms of asthma, course with neutrophilic airway inflammation, which contributes to progressive worsening of pulmonary damage and function by releasing: (i) elastase: cleaves elastin and causes bronchiectasis, decreases opsonization and phagocytosis, increases secretion, decrease mucociliary clearance; (ii) DNA: increases the viscosity of secretions; (iii) hydrogen peroxide and other oxidants: causes tissue damage and inactivates α-1-antitrypsin; (iv) IL-8 and LTB4: attract more neutrophils.5
Two aspects of COPDC have been thoroughly studied: the genetic component and environmental aggressions that initiate or exacerbate the diseases. Most COPDC show different genotypes, phenotypes, endotypes, and degrees of severity, require different types of management, and have no cure.6
Pneumonia, BPD, BO, and/or RWI in the first years of life constitute risk groups for COPD in the long term and should receive medical follow-up and interventions to prevent the potential impact on long-term respiratory sequelae.7–9
In all COPDC, PE is often triggered by viral and/or bacterial infections, pollution, and aeroallergens. The PE manifests as acute respiratory failure of varying intensity, both in asthma and RWI, and as increased cough and chronic infection in CF, BPD, NCFB, and PCD. The signs and symptoms of PE are more frequent and intense at nighttime.
While the majority of acute respiratory diseases can be diagnosed easily and efficiently through history and physical examination, those with a chronic nature may require sophisticated laboratory tests. Continuous and scheduled evaluations by interdisciplinary health teams in specialized centers are required for effective management, better prognosis, and improved quality of life in COPDC.
Establishing protocols based on systematic reviews, meta-analyses, and guidelines allows for gaining control of the signs and symptoms of COPDC. Oral and (OC) inhaled corticosteroids (IC), mucolytics, bronchodilators, inhaled antibiotics, and other drugs should be used according to specific guidelines for each COPDC.
While some groups of drugs are widely used, with variable degree of scientific evidence, such as bronchodilators and IC, others, such as long-term macrolide use, are more controversial. The use of macrolides in COPDC have been justified by the anti-inflammatory and immunomodulatory effect, the decrease in mucus production, and neutrophil elastase inhibition, in addition to reducing the production of pro-inflammatory mediators (e.g. IL8) and stimulating phagocytosis of apoptotic cells.10–14 Although macrolides have shown to be effective in patients with CF and other COPDC, and have shown encouraging in vitro effects, their use in other COPDC has shown to be less effective and requires further studies.14
The objective of this review was to assess and describe the main events related to the diagnosis and management of COPDC, considering the physiopathology and interrelated genetic and environmental characteristics.
Data sourcesReview of the PubMed databases with selection of relevant references. The following descriptors were used (according to the MeSH criteria) related to each COPD included in the study: asthma, CF, RWI, BPD, PCD, NCFB, BO, and PB.
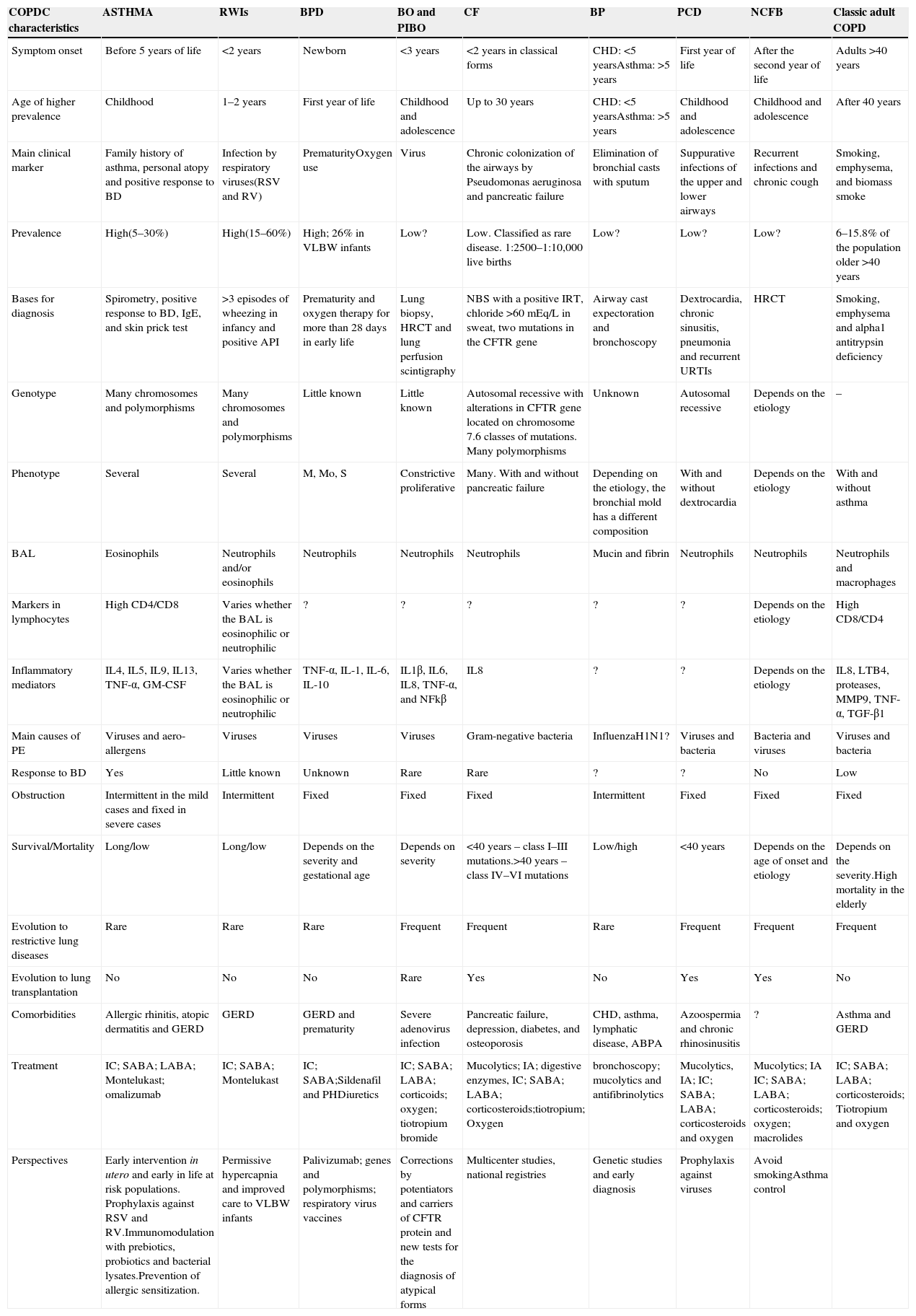
COPDC: an overviewThe first important feature of COPDC is that their clinical manifestations and prevalence are more common in certain age groups, as shown in Fig. 1. The second is that interactions between the genetic component and environmental aggressions initiate or exacerbate them, as shown in Fig. 2. The main differences and similarities between the COPDC are shown in Table 1. The third characteristic of COPDC is that they evolve with PE, predominantly infectious (CF), inflammatory (asthma), and mixed (asthma and viruses/bacteria).
 compared with chronic obstructive pulmonary disease (COPD) in adults. CF, cystic fibrosis; RWIs, recurrent wheezing in infants; BPD, bronchopulmonary dysplasia; PCD, primary ciliary dyskinesia; NCFB, non-cystic fibrosis bronchiectasis; PB, plastic bronchitis; BO, bronchiolitis obliterans; m, month; y, year.")
Age of onset and higher prevalence of major chronic obstructive pulmonary disease in children (COPDC) compared with chronic obstructive pulmonary disease (COPD) in adults. CF, cystic fibrosis; RWIs, recurrent wheezing in infants; BPD, bronchopulmonary dysplasia; PCD, primary ciliary dyskinesia; NCFB, non-cystic fibrosis bronchiectasis; PB, plastic bronchitis; BO, bronchiolitis obliterans; m, month; y, year.
.")
Main clinical and pathophysiological characteristics of COPDC versus COPD in adults.
| COPDC characteristics | ASTHMA | RWIs | BPD | BO and PIBO | CF | BP | PCD | NCFB | Classic adult COPD |
|---|---|---|---|---|---|---|---|---|---|
| Symptom onset | Before 5 years of life | <2 years | Newborn | <3 years | <2 years in classical forms | CHD: <5 yearsAsthma: >5 years | First year of life | After the second year of life | Adults >40 years |
| Age of higher prevalence | Childhood | 1–2 years | First year of life | Childhood and adolescence | Up to 30 years | CHD: <5 yearsAsthma: >5 years | Childhood and adolescence | Childhood and adolescence | After 40 years |
| Main clinical marker | Family history of asthma, personal atopy and positive response to BD | Infection by respiratory viruses(RSV and RV) | PrematurityOxygen use | Virus | Chronic colonization of the airways by Pseudomonas aeruginosa and pancreatic failure | Elimination of bronchial casts with sputum | Suppurative infections of the upper and lower airways | Recurrent infections and chronic cough | Smoking, emphysema, and biomass smoke |
| Prevalence | High(5–30%) | High(15–60%) | High; 26% in VLBW infants | Low? | Low. Classified as rare disease. 1:2500–1:10,000 live births | Low? | Low? | Low? | 6–15.8% of the population older >40 years |
| Bases for diagnosis | Spirometry, positive response to BD, IgE, and skin prick test | >3 episodes of wheezing in infancy and positive API | Prematurity and oxygen therapy for more than 28 days in early life | Lung biopsy, HRCT and lung perfusion scintigraphy | NBS with a positive IRT, chloride >60mEq/L in sweat, two mutations in the CFTR gene | Airway cast expectoration and bronchoscopy | Dextrocardia, chronic sinusitis, pneumonia and recurrent URTIs | HRCT | Smoking, emphysema and alpha1 antitrypsin deficiency |
| Genotype | Many chromosomes and polymorphisms | Many chromosomes and polymorphisms | Little known | Little known | Autosomal recessive with alterations in CFTR gene located on chromosome 7.6 classes of mutations. Many polymorphisms | Unknown | Autosomal recessive | Depends on the etiology | – |
| Phenotype | Several | Several | M, Mo, S | Constrictive proliferative | Many. With and without pancreatic failure | Depending on the etiology, the bronchial mold has a different composition | With and without dextrocardia | Depends on the etiology | With and without asthma |
| BAL | Eosinophils | Neutrophils and/or eosinophils | Neutrophils | Neutrophils | Neutrophils | Mucin and fibrin | Neutrophils | Neutrophils | Neutrophils and macrophages |
| Markers in lymphocytes | High CD4/CD8 | Varies whether the BAL is eosinophilic or neutrophilic | ? | ? | ? | ? | ? | Depends on the etiology | High CD8/CD4 |
| Inflammatory mediators | IL4, IL5, IL9, IL13, TNF-α, GM-CSF | Varies whether the BAL is eosinophilic or neutrophilic | TNF-α, IL-1, IL-6, IL-10 | IL1β, IL6, IL8, TNF-α, and NFkβ | IL8 | ? | ? | Depends on the etiology | IL8, LTB4, proteases, MMP9, TNF-α, TGF-β1 |
| Main causes of PE | Viruses and aero-allergens | Viruses | Viruses | Viruses | Gram-negative bacteria | InfluenzaH1N1? | Viruses and bacteria | Bacteria and viruses | Viruses and bacteria |
| Response to BD | Yes | Little known | Unknown | Rare | Rare | ? | ? | No | Low |
| Obstruction | Intermittent in the mild cases and fixed in severe cases | Intermittent | Fixed | Fixed | Fixed | Intermittent | Fixed | Fixed | Fixed |
| Survival/Mortality | Long/low | Long/low | Depends on the severity and gestational age | Depends on severity | <40 years – class I–III mutations.>40 years – class IV–VI mutations | Low/high | <40 years | Depends on the age of onset and etiology | Depends on the severity.High mortality in the elderly |
| Evolution to restrictive lung diseases | Rare | Rare | Rare | Frequent | Frequent | Rare | Frequent | Frequent | Frequent |
| Evolution to lung transplantation | No | No | No | Rare | Yes | No | Yes | Yes | No |
| Comorbidities | Allergic rhinitis, atopic dermatitis and GERD | GERD | GERD and prematurity | Severe adenovirus infection | Pancreatic failure, depression, diabetes, and osteoporosis | CHD, asthma, lymphatic disease, ABPA | Azoospermia and chronic rhinosinusitis | ? | Asthma and GERD |
| Treatment | IC; SABA; LABA; Montelukast; omalizumab | IC; SABA; Montelukast | IC; SABA;Sildenafil and PHDiuretics | IC; SABA; LABA; corticoids; oxygen; tiotropium bromide | Mucolytics; IA; digestive enzymes, IC; SABA; LABA; corticosteroids;tiotropium; Oxygen | bronchoscopy; mucolytics and antifibrinolytics | Mucolytics, IA; IC; SABA; LABA; corticosteroids and oxygen | Mucolytics; IA IC; SABA; LABA; corticosteroids; oxygen; macrolides | IC; SABA; LABA; corticosteroids; Tiotropium and oxygen |
| Perspectives | Early intervention in utero and early in life at risk populations. Prophylaxis against RSV and RV.Immunomodulation with prebiotics, probiotics and bacterial lysates.Prevention of allergic sensitization. | Permissive hypercapnia and improved care to VLBW infants | Palivizumab; genes and polymorphisms; respiratory virus vaccines | Corrections by potentiators and carriers of CFTR protein and new tests for the diagnosis of atypical forms | Multicenter studies, national registries | Genetic studies and early diagnosis | Prophylaxis against viruses | Avoid smokingAsthma control |
?, unknown; ABPA, allergic bronchopulmonary aspergillosis; IA, inhaled antibiotics; BD, bronchodilator; NCFB, non-cystic fibrosis bronchiectasis; BO, bronchiolitis obliterans; PIBO, post-infectious bronchiolitis obliterans; PB, plastic bronchitis; CHD, congenital heart disease; IC, inhaled corticosteroids; CD4, CD4 molecule; CD8, CD8 molecule; CFTR, cystic fibrosis transmembrane regulator; BPD, bronchopulmonary dysplasia; PCD, primary ciliary dyskinesia; COPD, chronic obstructive pulmonary disease; COPDC, chronic obstructive pulmonary disease in childhood; GERD, gastroesophageal reflux disease; PE, pulmonary exacerbation; CF, cystic fibrosis; GM-CSF, granulocyte macrophage-colony stimulating factor; PH, pulmonary hypertension; IgE immunoglobulin E; IL, interleukin; API, asthma predictor index; URTI, upper respiratory tract infection; LABA, long-acting beta-agonists; BAL, bronchoalveolar lavage; LTB4 leukotriene B4; M, mild; Mo, moderate; S, severe; MMP, matrix metallopeptidase; NF-kB, NF-kappaB; VLBW, very low birth weight; RSV, respiratory syncytial virus; RV, rhinovirus; SABA, short-acting beta agonists; RWI, recurrent wheezing in infants; HRCT, high-resolution computed tomography; SPT, skin prick test; TGF, transforming growth factor; IRT, immunoreactive trypsinogen; NBS, newborn screening test; TNF, tumor necrosis factor; RSV, respiratory syncytial virus.
Infectious PE caused by bacteria are different in healthy children when compared with those with COPDC. Streptococcus pneumoniae, Haemophilus influenzae and Staphylococcus aureus, which have high virulence, are common causes of pneumonia in previously healthy individuals without COPDC and require treatment with low-spectrum antibiotics (e.g., penicillin and amoxicillin), but cause short-term mortality. In contrast, many patients with COPDC, especially those with CF, PCD, and NCFB, have PE caused by low-virulence bacteria (Pseudomonas aeruginosa (PA), Burkholderia cepacia complex, Stenotrophomonas maltophilia and, Achromobacter xylosoxidans), which cause long-term mortality; however, they require broad-spectrum antibiotics such as aminoglycosides, meropenem, and third-generation cephalosporins (Fig. 3).
 and pneumonia in previously healthy children.")
Due to the characteristics of COPDC, several guidelines and systematic reviews for the diagnosis and management of the disease have been proposed for both periods between crises and PE of most COPDC, aiming to prevent further deterioration of pulmonary function and damage (Fig. 4).6,15–27
Asthma based on diagnosis.")
Asthmatic individuals who manifested the disease early in life persist with symptoms, and evidence suggests that asthma severity in childhood predicts the disease severity in adulthood.28 Recent guidelines emphasize difficulties for the definitive diagnosis of asthma in children younger than 5 years, where several COPDC phenotypes have been identified.
For children older than 5 years, asthma diagnosis is based on: (1) a history of acute respiratory failure crises that improve with short-acting bronchodilators (SABA); (2) increased serum IgE in the absence of parasitic diseases, eosinophilia, and positive immediate hypersensitivity skin tests for airborne allergens; (3) spirometry and measurement of bronchial hyperresponsiveness (BHR) to methacholine challenge. The diagnosis can be made with the isolated presence and/or combination of the above items 1; 1+2; 1+3; or 1+2+3.
Numerous factors have been associated with increased risk of developing asthma in adolescence, of which the following should be mentioned: personal or family history of allergy, male gender, obesity, pollution, exposure to cigarette smoke, RWI, severe infection by respiratory syncytial virus (RSV), pneumonia in the first year of life, RWI severity, altered pulmonary function, and BHR.8,29–31
There is no specific treatment for asthma, and it is preferable to use the term management for the tools used to attain disease control. The main objectives for outpatient management are: (i) make chronic symptoms minimal or nonexistent; (ii) decrease the intensity and the number of PE; (iii) maintain lung function as close as possible to normal levels; (iv) maintain normal levels of daily activities, including physical exercises; (v) prevent the adverse effects of anti-asthmatic drugs; (vi) prevent disease progression to irreversible airflow limitation; (vii) prevent asthma mortality.6,15–20 The main medications to control asthma are inhaled corticosteroids (IC).
The classification of the clinical pictures and respective treatment steps are listed below:
Steps in the treatment of asthma(Step 1) Individuals with intermittent asthma, characterized by normal spirometry and periods between crises lasting longer than 1 month without exacerbations in the last year, should receive SABA. (Step 2) Continued use of IC at low doses and rescue SABA during crisis. (Step 3) Continued use of low doses of IC+long-acting beta agonists (LABA) with SABA for crises or IC+formoterol for both maintenance and control. (Step 4) IC+formoterol for maintenance and crises or medium or high doses of IC+LABA and SABA, if necessary. (Step 5) Refer patient to a center specialized in asthma treatment for difficult-to-control disease: consider oral steroids and anti-IgE (omalizumab). Consider oral corticosteroids in severe PE and if the patient had previous severe PE.6,15–20
Medications and steps are modified to the next step or revert to the previous step, depending on whether or not the asthma is controlled. A decrease in management steps must occur if the asthma is well controlled for at least 3 months.
It is essential to verify at all consultations, whether the asthma is controlled or not, from the clinical and/or functional point of view, based on six parameters: (i) nocturnal signs and symptoms; (ii) daytime signals and symptoms; (iii) signs and symptoms with physical exercise or limitation in daily activities; (iv) PE; (v) need for relief medication (SABA); (vi) changes in lung function. Hence, asthma is considered controlled when all parameters are normal; it should be classified as partially controlled in the presence of one or two altered parameters; and finally, non-controlled asthma should be considered when three or more parameters are altered.
The absence of control of signs and symptoms, frequent exacerbations, previous admission to an intensive care unit (ICU), low values of forced expiratory volume in 1s (FEV1), exposure to tobacco smoke, and need to use high-dose medications are characteristics associated with increased risk of adverse events in the future. By definition, an exacerbation at any week is indicative of non-controlled asthma and also of the need for review of the maintenance treatment.16,17
In the management of patients with asthma, the following are essential: (i) management supported by evidence-based medicine; (ii) to perform the diagnosis and, if possible, the phenotype (e.g., allergic and non-allergic); (iii) to exclude and treat comorbidities; (iv) to assess and recommend the adequate use of prescribed drugs; (v) to assess, advise, and encourage treatment adherence; (vi) to assess and advise about environmental prophylaxis; (vii) to assess and advise on the triggering factors; (viii) to educate patient's caregivers about asthma and the factors influencing it; (ix) to give instructions on the adequate use of devices for administration of metered-dose and dry powder inhalers; (x) instructions for patients to be able to recognize when asthma control is deteriorating and what medications to use, when it occurs; (xi) to identify non-controlled patients and causes of lack of control; (xii) to advise that inhaled medications should be used with spacers; (xiii) to advise on the hygiene of spacers, which must be washed and left to soak in water with detergent; (xiv) LABA must not be used in children younger than 4 years; (xv) SABA are the agents of choice in PE; (xvi) IC alone or associated with bronchodilators are the basis of asthma treatment; (xvii) children younger than 6 years can use inhaled medication with spacer and those older than 6 years can use dry powder inhalers; (xviii) to assess pulmonary function regularly; (xix) to advise on the need for long-term medical care; (xx) omalizumab should be prescribed in reference centers for the management of patients with difficult-to-control asthma.
Recurrent wheezing in infants (RWI)There are several phenotypes and risk factors (RF) for RWI, creating difficulties for asthma diagnosis and resulting in an excessive assessment for comorbidities.32–37 The main RF include: presence of familial and/or personal allergy, early sensitization, severe RSV infection, maternal smoking during pregnancy, and unfavorable airway geometry. Other implicated RF are: genetic variants, excessive hygiene, Western lifestyle, pollution, gastroesophageal reflux disease (GERD), low socioeconomic status, urban environment, antibiotic use, diet, few siblings, ethnicity, male gender, and attendance of daycare.38
Exposure to maternal smoking, both intrauterine and in the early years of life, has contributed to increased incidence and severity of childhood and adult asthma, COPD in adults, lung function deficits, lung hypoplasia, respiratory tract infections, and higher predisposition to sudden death syndrome.39–41 Respiratory viruses are the main RF for asthma and RWI initiation or exacerbation: (i) some viruses initiate asthma; (ii) the more severe the respiratory infection, the higher the likelihood of developing asthma; (iii) viruses cause PE in children and adults with established asthma.42
The diagnosis of asthma in infants is a major challenge for clinicians; to attain a diagnosis with a high degree of suspicion, the presence of allergy should be investigated, as it is a persistent asthma predictor. The investigation of severe COPDC in children younger than 5 years may require many tests, including high-resolution computed tomography (HRCT), lung function assessment, cellular and humoral immunity evaluation, bronchoscopy, bronchoalveolar lavage, 24-h esophageal pH-metry, endobronchial biopsy, mutation screening, and sodium and chloride sweat measurements.35 In contrast, no investigation is necessary for most of RWI, because it cannot differentiated with certainty whether the future course of the RWI will be persistent asthma or transient wheezing.43
Some asthma predictive indices (APIs) have been developed to identify children with RWI and at risk for asthma after 6 years of age (Table 2).38,44 Some authors have mentioned methodological and practical limitations of APIs, suggesting that they have low capacity and poor sensitivity to predict asthma at school age,45,46 and that although they are simple and easy-to-apply tools, they have not been sufficiently validated. According to these authors, predicting asthma using simple clinical models is virtually impossible.45,46
| API criteria | Original API1 | Modified API38 |
|---|---|---|
| General | Convincing API: more than three episodes of wheezing/year during the first 3 years of life with a major criterion or two minor. | The child must have a history of four or more episodes of wheezing. At least one episode verified and confirmed by a physician. |
| Less convincing API: less than three episodes of wheezing/year during the first 3 years of life with a major criterion or two minor. | ||
| Major | History of parental asthmaAtopic dermatitis | History of parental asthmaAtopic dermatitis diagnosed by physicianAllergic sensitization to one or more aeroallergens |
| Minor | Allergic rhinitis diagnosed by physicianWheezing unrelated to coldsEosinophilia≥4% | Sensitization to cow's milk, egg, or peanutWheezing unrelated to coldsEosinophilia≥4% |
API, asthma predictive index.
Although most of the scientific community is favorable to the use of APIs,47,48 the search for clinical and/or laboratory markers for the diagnosis of asthma in RWI and children younger than 5 years remains a rich area for future studies. Infants and preschoolers with RWI or asthma have less PE, and show symptom and lung function improvement when treated with IC.49–51
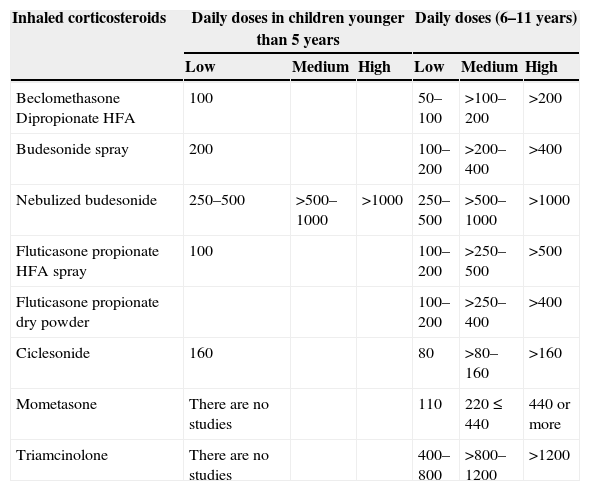
The dosages of the main IC used in COPDC are shown in Table 3.
Low, medium, and high daily doses of inhaled corticosteroids (IC) in asthma (Global Initiative for Asthma – GINA) and in chronic obstructive pulmonary disease in childhood.
| Inhaled corticosteroids | Daily doses in children younger than 5 years | Daily doses (6–11 years) | ||||
|---|---|---|---|---|---|---|
| Low | Medium | High | Low | Medium | High | |
| Beclomethasone Dipropionate HFA | 100 | 50–100 | >100–200 | >200 | ||
| Budesonide spray | 200 | 100–200 | >200–400 | >400 | ||
| Nebulized budesonide | 250–500 | >500–1000 | >1000 | 250–500 | >500–1000 | >1000 |
| Fluticasone propionate HFA spray | 100 | 100–200 | >250–500 | >500 | ||
| Fluticasone propionate dry powder | 100–200 | >250–400 | >400 | |||
| Ciclesonide | 160 | 80 | >80–160 | >160 | ||
| Mometasone | There are no studies | 110 | 220≤440 | 440 or more | ||
| Triamcinolone | There are no studies | 400–800 | >800–1200 | >1200 | ||
Bronchiolitis obliterans (BO) is a predominantly neutrophilic COPDC with high levels of proinflammatory cytokines, bronchial remodeling, and fibrosis in the small airways. When the etiology is infectious, it is called post-infectious BO (PIBO). Most often, PIBO is caused by previous infection caused by adenovirus, but influenza, measles, RSV, and Mycoplasma pneumoniae are also observed. Latin American countries have the highest rates of this disease.27,52–62
Studies have shown that lung function remains altered with an obstructive pattern and air trapping during childhood. These patients are often hospitalized due to recurrent respiratory infections. Patients show slight improvement over the years.54,62
Five criteria are essential for the diagnosis of PIBO: (i) history of acute viral bronchiolitis and viral pneumonia before the age of 3; (ii) evidence of persistent airway obstruction after the acute event, identified by physical examination and/or lung function, which is not responsive to at least 2 weeks of systemic corticosteroids associated with bronchodilator; (iii) radiological findings of obstruction such as hyperinflation, atelectasis, bronchial wall thickening, and bronchiectasis; (iv) mosaic perfusion and air trapping on HRCT; (v) exclusion of other COPDC.27,60
Among the causes of BO are: drugs, association with Stevens–Johnson syndrome, collagenoses, irradiation, foreign body or gastric content aspiration, and graft vs. host disease after transplantation.
The definitive diagnosis of BO is made through anatomopathological examination of fragments obtained from lung biopsy. BO is classified as: (i) proliferative BO, characterized by airway obstruction by polyps and/or granulation tissue in the lumen of bronchioles; or (ii) constrictive BO, characterized by peribronchial inflammation and fibrosis.58 As the lung biopsy is an invasive test, most studies perform the diagnosis based on clinical history and HRCT, spirometry, and perfusion scintigraphy with technetium. Alterations in HRCT include wall thickening and bronchiole obliteration and, with the disease progression, bronchiectasis, bronchial wall thickening, mosaic perfusion, air trapping, reduced lung volume, and decreased diameter of hilar and peripheral vessels are observed.27,59
There is no specific treatment for BO. Oxygen therapy is essential in some patients, mainly after ICU discharge for severe acute viral bronchiolitis (AVB) and in patients who develop chronic hypoxemia. Contact prophylaxis with intra and extra-domestic pollutants, reduction of exposure to active and passive smoking, and anti-pneumococcal and influenza vaccination are important steps for BO management. In the early phases of the disease, some patients may benefit from systemic corticosteroid therapy. Bronchodilators, antibiotics in infectious exacerbations, surgery for resection of fixed collapsed lung, and lung transplant have also been used.27
Primary ciliary dyskinesia (PCD)Primary ciliary dyskinesia (PCD) is an autosomal recessive COPDC caused by several alterations in airway cilia anatomical and functional structure, resulting in inflammation and infection, with an incidence of 1:4000–40,000 live births. Laterality defects in the thoraco-abdominal organs occur in about 50% of patients with PCD, and most have situs inversus totalis (Kartagener syndrome).24,63–66
PCD should be suspected in children with: (i) situs inversus totalis or other laterality defect in thoraco-abdominal organs; (ii) recurrent upper (otitis, sinusitis) and lower respiratory tract infections (pneumonia, abscesses) and chronic respiratory tract diseases of undefined etiology (bronchiectasis, chronic cough, atelectasis, and middle lobe syndrome); (iii) neonatal respiratory distress syndrome of undefined causes; (iv) diagnosis of PCD in other family members; (v) congenital heart disease, especially if associated with heterotaxia, and with chronic and repeated infections of the lower and upper airways.24,67
Clinical manifestations and alterations in PCD vary according to age25: in the prenatal period, ultrasound may reveal mild fetal cerebral ventriculomegaly, heterotaxia and situs inversus totalis (approximately 25% of individuals with situs inversus totalis have PCD). PCD prevalence in patients with heterotaxia remains unknown. In the neonatal period, 75% of full-term newborns with PCD have respiratory distress, requiring supplemental oxygen for days or weeks. Some have continuous rhinorrhea, heterotaxia, and hydrocephalus.
During childhood, chronic productive cough is observed, in association with atelectasis and/or recurrent pneumonia, atypical asthma that does not respond to treatment, bronchiectasis, NCFB, nasal polyps, chronic rhinosinusitis, hearing impairment, and chronic otitis. In adolescence and adulthood, bronchiectasis, chronic mucopurulent sputum, digital clubbing, spirometry with progressive or mixed obstructive ventilatory pattern, nasal polyposis, and halitosis can be observed, as well as infertility in men (50%) and ectopic pregnancy in women.
Diagnosis is based on clinical and radiological alterations, nasal nitric oxide levels, ciliary beat pattern at video microscopy, structural alterations of the cilia by electron microscopy, and detection of mutations. Only 50–60% of PCD patients have a known mutation.25,68,69
CF evolves with secondary ciliary dyskinesia, whereas in PCD it is primary; the management of pulmonary disease in these two diseases is virtually identical.25
Cystic fibrosis (CF)CF is an autosomal recessive disease caused by mutations in the CFTR (cystic fibrosis transmembrane regulator) gene, 7q31.2 region, which has more than 2000 identified mutations, divided into six classes regarding the production and function of the CFTR protein. The qualitative or quantitative absence or dysfunction of CFTR causes physiopathological changes in several organs. Most of CF morbidity and mortality is caused by manifestations in the respiratory and digestive tracts.70–73
It is currently known that in Class I, II, and III mutations, the disease starts before symptom onset, constituting a major factor for early intervention and more aggressive treatments, showing promising results in reducing lung function damage and deterioration, with consequent increased survival.70–73
Availability of newborn screening, sweat testing, CFTR gene mutation screening, medications, and possible use of correctors, enhancers, and stabilizers (for some mutations) of the CFTR protein are concrete facts in the last decade. The consequence is that the survival of these patients is increasingly higher.73 Lung damage and decreased lung function are progressive, and depend on the mutation, gender, polymorphisms, treatment availability and adherence, early treatment, comorbidities, and care in reference centers.74–76
The physiopathology of COPD in CF can be understood with the following six events (6D): (i) defect in the CFTR gene; (ii) impairment or absence of the CFTR protein; (iii) dehydration of the airway surface liquid; (iv) defect in bacterial clearance in the airways; (v) secondary ciliary dyskinesia; (vi) destruction of the airway epithelium.77
To attain the diagnosis of CF after neonatal screening, alterations in the gene and or CFTR protein should be verified. The CFTR gene is studied through the analysis of mutations and polymorphisms. The CFTR protein function and/or presence can be assessed in the sweat glands (sweat test, evaporimetry, difference of potential, and pH) nasal epithelium, salivary glands, and digestive tract.78,79
Diagnosing or ruling out CF is not always easy, even after comprehensive testing, particularly in adolescents and adults with classes IV, V, and VI mutations, which cause non-classical forms and are often associated with rare mutations, threshold levels of chloride in sweat, late-onset clinical manifestations, and atypical symptoms.80
As CF affects many organs, its management is complex. The service must have interdisciplinary teams with several health care professionals, physical therapy, medical examinations, frequent visits to the hospital, and use of daily, repetitive medications, which are sometimes complicated and costly.81
Patients with CF are highly susceptible to chronic lung infection by PA. PA acquisition occurs very early and is related to lung function deterioration and worse CF prognosis; hence, intensive treatment against PA is crucial in the management of CF.
Other bacteria frequently found in the airways of CF patients and that determine the lung function damage and deterioration include S. aureus, H. influenzae, PA, B. cepacia complex, S. maltophilia, and Achromobacter species. Non-tuberculous Mycobacteria (NTM) and other anaerobic bacteria and fungi are found less frequently, and tend to appear at the more advanced stages of lung disease.82,83
Inhaled antibiotics (IA) are essential for the treatment of chronic lung infections in CF. The increased survival in patients with CF in recent decades can be partly attributed to the use of IA. They have higher deposition at the infection site and less risk of systemic side effects than parenteral therapy, decrease PE, and improve quality of life and spirometry.84,85
CF patients with chronic lung infection by PA should receive long-term, nebulized anti-pseudomonal therapy, unless there is contraindication. Among the IA, tobramycin and colistin are currently available, while aztreonam, levofloxacin, and liposomal amikacin will soon be available.86 The administration of IA is time-consuming and is a heavy burden for patients. Treatment adherence is a significant challenge.87
The treatment of the first acquisition episode, from infection to chronic infection and PE due to PA, are reasons for debates and controversies in the literature.88
PA eradication protocols have demonstrated antibiotic efficacy in the first bacterial isolation; intensive treatment with intravenous antibiotics in PE and IA in the periods between crises.89–91
It is advisable to perform lung damage assessment by HRCT at least every 2 years, and pulmonary function assessment by spirometry at least twice a year.
IA, anti-inflammatory drugs, corticosteroids, bronchodilators, and mucolytics may be necessary for the management of lung disease in CF. Auxiliary measures have shown benefits with respiratory physical therapy, regular exercise, avoiding indoor and outdoor pollution, and active and passive smoking. It is crucial to include treatment adherence measures and continuity of care for life. Visits to the reference center must be monthly during the first year and at least four times a year until adulthood.
Adherence to CF management varies between 35 and 75% and is lower in adolescence. Poor adherence to medication worsens lung function, increases the frequency of PE, and results in a larger number of consultations, hospitalizations, and costs. Therefore, interventions are necessary to improve medication adherence.81
Many international organizations have proposed that the management of lung disease in CF should be directed by guidelines based on scientific evidence. Thus, recent publications on the management of COPD in CF can be found, and represent the “state-of-the-art” for CF reference centers.82,83,91,92
Recently, advances in the treatment of CF have gained prominence. Mutations in the CFTR gene have been widely identified, and the highest prevalence of the F508del mutation has been emphasized. Treatment for the specific mutation and/or class of mutations has focused on the study of new drugs. Two drugs have been used in studies considering the F508del mutation in homozygous form, and results of the association of ivacaftor (increases opening of the chloride channel – potentiator) and lumacaftor (increases protein number in the epithelium – potentiator) have shown increased chloride transport when compared with the values of the individual drugs.93
Bronchopulmonary dysplasia (BPD)BPD is a secondary COPDC, and is associated with prematurity and risk factors (RF) related to it. Low birth weight, high fraction of inspired oxygen, patent ductus arteriosus, high intravenous fluid volume values, mechanical ventilation with high pressures, genetic susceptibility, and infection in the neonatal period contribute to the prevalence and severity of BPD. This disease affects approximately 30% of newborns with birth weight less than 1500g.94,95
Several studies have shown that most infants who develop BPD evolve with lung function alterations throughout life.9,96–101
Similar to all COPDC, BPD has no specific treatment. Regarding the advances to reduce mortality in the neonatal period, in recent decades, three factors have clearly proven to be effective: (i) corticosteroid administration to pregnant women with high-risk pregnancies; (ii) administration of surfactants to premature infants; (iii) less aggressive ventilation strategies.102,103
The following have also been used: diuretics (furosemide); bronchodilators; systemic corticosteroids and IC; vitamin A; methylxanthines (pentoxifylline, caffeine); pulmonary vasodilators (sildenafil, inhaled nitric oxide); late surfactant administration; and antioxidants (superoxide dismutase), with varying degrees of efficacy in the management of acute and/or chronic BPD. However, there is little evidence of the beneficial actions of most of these agents when used in short- or long-term management of BPD.102,103
BPD is associated with a high incidence of pulmonary arterial hypertension (PAH). Treatment with sildenafil has been associated with significant improvement in echocardiographic markers of PAH and reduced need for oxygen. The drug is well tolerated.104
Bronchiectasis related to diseases other than CF (NCFB)Bronchiectasis is an alteration that accompanies many COPDC. It may be associated with and complicate asthma, PCD, CF, BO, and NCFB, and be consequent to measles, pertussis, immunodeficiency, allergic bronchopulmonary aspergillosis, inflammatory bowel disease, rheumatoid arthritis, foreign body inhalation, PCD, BO, respiratory tract malformation (e.g.: tracheoesophageal fistula, congenital cystic adenomatoid malformation) and result from inflammatory and infectious diseases to the airways. NCFB encompasses several etiologies and degrees of severity; a clear cause cannot be established in many patients.105,106
Pulmonary disease in NCFB is very similar to CF, BO, and PCD. The challenge a child with NCFB faces is the pursuit of a diagnosis. The management is very similar to that of most COPDC and should be considered on a case-by-case basis, aiming at patient comfort and clinical response.107
Although some randomized, controlled, double-blind trials have shown that prolonged use of macrolides in patients with NCFB improves quality of life and reduces PE rates, the role of macrolides in NCFB management remains unclear and they cannot be recommended for routine use. Until very recently, these drugs were investigated in small trials of short duration, which did not evaluate relevant clinical outcomes, such as PE and quality of life.
Long-term oral antibiotics should not be routinely prescribed. Macrolides (or other antibiotics) can be considered for a limited period of 12–24 months, especially in patients with frequent exacerbations.4,108,109
Plastic bronchitis (PB)PB is a rare COPDC with unknown prevalence and etiology, with formation of bronchial molds of gelatinous or solid appearance in the large airways, whose composition varies, consisting of mucins, fibrin, DNA, or eosinophils, depending on the PB etiology. The diseases most often associated with PB are congenital heart disease and asthma. It has also been described in infection by influenza virus, H1N1, lymphatic diseases, allergic bronchopulmonary aspergillosis, and sickle-cell disease.109 Although bronchodilators, corticosteroids, antibiotics, mucolytics, physical therapy, antifibrinolytic therapy with heparin, and urokinase have been used, single or repeat bronchoscopy is essential for diagnosis and treatment.
Final considerationsThe pediatrician's actions are vital for the management of COPDC. The clinician should suspect the disease in the presence of signs and symptoms in order to attain an early and accurate diagnosis, know the risk factors and associated comorbidities, and assess treatment adherence, the correct use of prescribed drugs, and their side effects based on well-defined management protocols, preferably based on specific guidelines.
Management benefits should be assessed through the reduction in acute pulmonary exacerbations, increased quality of life, and decreased evolution of lung function loss (spirometry, measurement of oxygen saturation, lung clearance index) and lung damage (HRCT every 2 years, if necessary). Therefore, the sequential evaluation of lung function and damage should be standardized and continuous, throughout life. For most COPDC, monitoring in specialized reference centers by interdisciplinary teams leads to better outcomes.
Conflicts of interestThe authors declare no conflicts of interest.
To Professor Dr. Paulo Augusto Moreira Camargos for his encouragement, suggestions, and manuscript review. To Professor Dr. Fernando Augusto de Lima Marson, for the excellent chart contributions, text reading, suggestions, and appraisals. To the Pediatric Pneumology Team of Universidade Estadual de Campinas (Unicamp), for their constant help in the management of COPDC for many years: Adyleia Aparecida Dalbo Contrera Toro, Maria Angela Gonçalves de Oliveira Ribeiro, Andressa Peixoto, Milena Baptistela Grotta, Emilia Gonçalves, and Maria Cristina Simões Ferreira.
Please cite this article as: Ribeiro JD, Fischer GB. Chronic obstructive pulmonary diseases in children. J Pediatr (Rio J). 2015;91:S11–25.




