


Arthrogryposis multiplex congenita (AMC) is a relatively rare neuromuscular syndrome, with a prevalence of 1:3000–5000 newborns. In this study, the authors describe the clinical features of a group of 50 unrelated Mexican patients with arthrogryposis multiplex congenita.
MethodsPatients were diagnosed by physical and radiographic examination and the family history was evaluated.
ResultsOf the 50 cases, nine presented other features (pectum excavatum, cleft palate, mental retardation, ulnar agenesis, etc.). Environmental factors, as well as prenatal and family history, were analyzed. The chromosomal anomalies and clinical entities associated with arthrogryposis multiplex congenita were reported. No chromosomal aberrations were present in the cases with mental retardation. Three unrelated familial cases with arthrogryposis multiplex congenita were observed in which autosomal recessive, autosomal dominant and X‐linked inheritance patterns are possible. A literature review regarding arthrogryposis multiplex congenita was also conducted.
ConclusionsIt is important to establish patient‐specific physical therapy and rehabilitation programs. A multidisciplinary approach is necessary, with medical, surgical, rehabilitation, social and psychological care, including genetic counseling.
A artrogripose múltipla congênita é uma síndrome neuromuscular relativamente rara, com prevalência de 1:3000‐5000 recém‐nascidos. É por isso que, neste estudo, descrevemos as características clínicas de um grupo de 50 casos de pacientes mexicanos não relacionados com artrogripose múltipla congênita.
MétodosOs pacientes foram diagnosticados por exame físico e radiográfico e o histórico familiar foi avaliado.
ResultadosDescrevemos 50 pacientes não relacionados com artrogripose múltipla congênita. Nove deles apresentaram outras características (pectus excavatum, fissura palatina, retardo mental, agenesia da ulna etc.). Foram analisados os fatores ambientais, pré‐natais e o histórico familiar. Relatamos as anomalias cromossômicas e as entidades clínicas associadas com a artrogripose múltipla congênita. Não havia aberração cromossômica nos casos com retardo mental. Também encontramos três casos familiares não relacionados com artrogripose múltipla congênita, em que são possíveis padrões de herança autossômica recessiva, autossômica dominante e ligada ao cromossomo X. Também analisamos a preocupação da literatura com a artrogripose múltipla congênita.
ConclusõesReiteramos a ideia de que é importante estabelecer programas de fisioterapia e reabilitação específicos para os pacientes. É necessária uma abordagem multidisciplinar com cuidado médico, cirúrgico, de reabilitação, social e psicológico, incluindo aconselhamento genético.
O termo artrogripose deriva das palavras gregas “arthron” e “gryposis” e significa “articulações encurvadas”. Vários sinônimos foram usados para descrever essa característica clínica: rigidez articular congênita múltipla, amioplasia congênita, miodistrofia fetal deformante, artromiodisplasia congênita, contraturas congênitas múltiplas e artrogripose múltipla congênita (AMC).1,2 A AMC refere‐se a um grande grupo heterogêneo de doenças, caracterizado por músculos contraídos e pela limitação congênita não progressiva do movimento de duas ou mais articulações diferentes com grossas cápsulas periarticulares. A AMC é relativamente rara, ocorre em aproximadamente 1:3000‐5000 recém‐nascidos e meninos e meninas são igualmente afetados. Todas as quatro extremidades estão envolvidas em 50‐60% dos casos, os membros inferiores em 30‐40% e os membros superiores em 10‐15%. Em 20% dos casos ocorre a luxação e subluxação dos quadris, das patelas e dos joelhos, ao passo que as articulações vertebrais e temporomandibulares raramente estão envolvidas.3,4 O tipo mais comum de artrogripose é a amioplasia, que representa um terço de todos os casos. A amioplasia é caracterizada pelo posicionamento simétrico dos membros com pé torto equinovaro grave e cotovelos estendidos. Uma característica que normalmente poderá ocorrer é um hemangioma facial médio característico. Alguns pacientes também podem apresentar anomalias estruturais ou genitais, os ombros são curvados e voltados para dentro e os antebraços são pronados com deformidades de flexão de punhos e dedos. Além do pé torto equinovaro, o envolvimento dos membros inferiores inclui frequentemente joelhos flexionados ou estendidos; os quadris poderão estar flexionados e voltados para fora ou estendidos e subluxados ou luxados.3,5,6
A AMC pode estar presente em mais de 150 doenças específicas e essas doenças incluem um grande grupo de doenças miopáticas e neurogênicas, anomalias no tecido conjuntivo e fatores que produzem uma limitação intrauterina do movimento fetal (anomalias estruturais do útero, síndrome da banda amniótica, entrelaçamento, oligodrâmnio ou polidrâmnio).7,8 Em modelos animais, as contraturas foram induzidas por vírus, bloqueadores neuromusculares, inseticidas, anticonvulsivantes, agentes bloqueadores neuromusculares, etanol, imobilização dos membros e hipertermia.8‐14 Esses fatores parecem interferir no desenvolvimento dos membros, resultam na perda de massa muscular com o desequilíbrio da força muscular nas articulações. A etiologia da artrogripose inclui tanto fatores genéticos quanto ambientais; entre as causas genéticas foram relatados defeitos em um único gene e cromossomopatias. A análise molecular revelou que um determinado fenótipo pode ser causado por mutações em diferentes genes, o que comprova a heterogeneidade genética dessa doença.3
O objetivo deste estudo é descrever as características clínicas de 50 casos de AMC e incluir uma breve análise da literatura a respeito desse assunto.
Paciente e métodosFoi feito um estudo prospectivo e transversal que incluiu uma série de 50 pacientes com AMC. Esses pacientes foram encaminhados ao Departamento de Genética do Instituto Nacional de Reabilitação (Cidade do México, México) com diagnóstico presumível de AMC durante um período de três anos. Todos eram descendentes de mexicanos. Os pacientes e seus pais foram informados sobre as características do estudo e concordaram em participar.
O protocolo foi aprovado pelos comitês de pesquisa e ética do Instituto. O diagnóstico de AMC teve como base características clínicas típicas, como contraturas articulares não progressivas que limitam movimentos de extensão ou de flexão evidentes no nascimento e envolvem mais de uma articulação, tanto dos membros superiores quanto dos inferiores ou de ambos. Em todos os casos, os pacientes foram submetidos a exame físico e radiográfico. Avaliamos o histórico familiar médico e as características do parto durante a consulta médica no Departamento de Genética. A avaliação de histórico de gravidez das mães dos pacientes incluiu a posição do feto no útero, infecções, febre, substâncias ilegais, medicamentos e acontecimentos incomuns durante a gravidez. A análise do cariótipo foi feita a partir dos leucócitos do sangue periférico com o uso da coloração banda G segundo técnicas padrão: resumidamente, o sangue periférico coletado por punção venosa, com o uso de uma seringa heparinizada, foi cultivado na presença de fito‐hemaglutinina. Após 72 horas, o crescimento das células foi interrompido ao se acrescentar demecolcina e as culturas de células foram colhidas e tratadas com solução hipotônica e fixadora. As amostras foram colocadas em lâminas de vidro para coloração.15 Foram analisadas 25 metáfases no microscópio de luz em cada caso.
ResultadosNo presente estudo, 28 (56%) dos pacientes eram do sexo feminino e 22 (44%) do masculino e ambos os sexos foram afetados. Apresentaram afecção das quatro extremidades 27 (54%) pacientes e tanto os membros inferiores quanto os superiores foram afetados em 18 (36%) e cinco (10%) casos, respectivamente. A distribuição de envolvimento dos membros é apresentada na figura 1A. O comprometimento articular pode apresentar uma grande variabilidade em um mesmo paciente: ao mesmo tempo em que uma articulação pode estar completamente estendida, a articulação contralateral pode estar flexionada. A figura 1B retrata o envolvimento clínico de membros superiores: a pronossupinação do antebraço foi comprometida bilateralmente, os pulsos e articulações interfalangeanas estavam fixados em posição semiflexionada e não apareceu linha de flexão. A figura 1C exemplifica o envolvimento dos membros inferiores: os joelhos direito e esquerdo foram fixados em posição semiflexionada e estendida, respectivamente, o tornozelo direito foi estendido e o tornozelo esquerdo em adução e a paciente não conseguia mover seus tornozelos. A idade média de apresentação na clínica foi de 4,6 anos (faixa etária de recém‐nascido a sete anos). Com relação ao envolvimento de outras articulações, observou‐se luxação e subluxação dos quadris em 14 casos (28%) e hipermobilidade articular com hiperflexão em dois casos. Uma grande variedade de anomalias congênitas (ortopédicas ou não ortopédicas) foi relatada como associada a AMC; em nossos pacientes, outras características clínicas eventuais também foram observadas em nove de 50 casos (tabela 1). Em nossa série, havia doença materna em 10 casos: um caso com diabetes mellitus, cinco casos apresentaram infecção do trato urinário e febre (acima de 39°C) no primeiro e segundo trimestre de gravidez, quatro casos apresentaram eclâmpsia e seis tiveram sangramento vaginal durante a gravidez no primeiro trimestre. O histórico dos partos em nossos pacientes mostrou que a posição do feto no parto foi cefálica em 25 casos, pélvica em 16, transversal em sete casos e outra posição em dois. Também foram observados um parto múltiplo (gêmeos dizigóticos) e um caso com anomalias no útero.
 Percentual de envolvimento dos membros. B) Membros superiores de um paciente afetado: os movimentos de pronossupinação do antebraço não eram possíveis, os pulsos e as articulações interfalangeanas estavam fixados em posição semiflexionada e não apareceu linha de flexão. C) Membros inferiores de um paciente com AMC: os joelhos direito e esquerdo foram fixados em posição semiflexionada e estendida, respectivamente, o tornozelo direito foi estendido e o tornozelo esquerdo em adução.")
Características gerais dos pacientes com artrogripose múltipla congênita estudados. A) Percentual de envolvimento dos membros. B) Membros superiores de um paciente afetado: os movimentos de pronossupinação do antebraço não eram possíveis, os pulsos e as articulações interfalangeanas estavam fixados em posição semiflexionada e não apareceu linha de flexão. C) Membros inferiores de um paciente com AMC: os joelhos direito e esquerdo foram fixados em posição semiflexionada e estendida, respectivamente, o tornozelo direito foi estendido e o tornozelo esquerdo em adução.
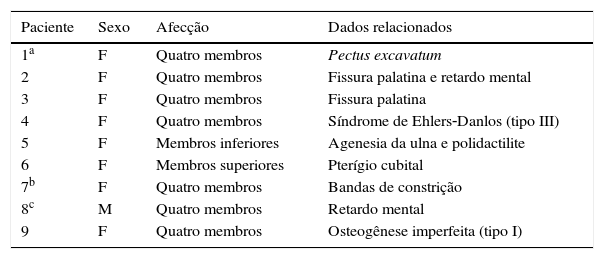
Anomalias congênitas associadas a artrogripose múltipla congênita em nossos pacientes
| Paciente | Sexo | Afecção | Dados relacionados |
|---|---|---|---|
| 1a | F | Quatro membros | Pectus excavatum |
| 2 | F | Quatro membros | Fissura palatina e retardo mental |
| 3 | F | Quatro membros | Fissura palatina |
| 4 | F | Quatro membros | Síndrome de Ehlers‐Danlos (tipo III) |
| 5 | F | Membros inferiores | Agenesia da ulna e polidactilite |
| 6 | F | Membros superiores | Pterígio cubital |
| 7b | F | Quatro membros | Bandas de constrição |
| 8c | M | Quatro membros | Retardo mental |
| 9 | F | Quatro membros | Osteogênese imperfeita (tipo I) |
F, sexo feminino; M, sexo masculino.
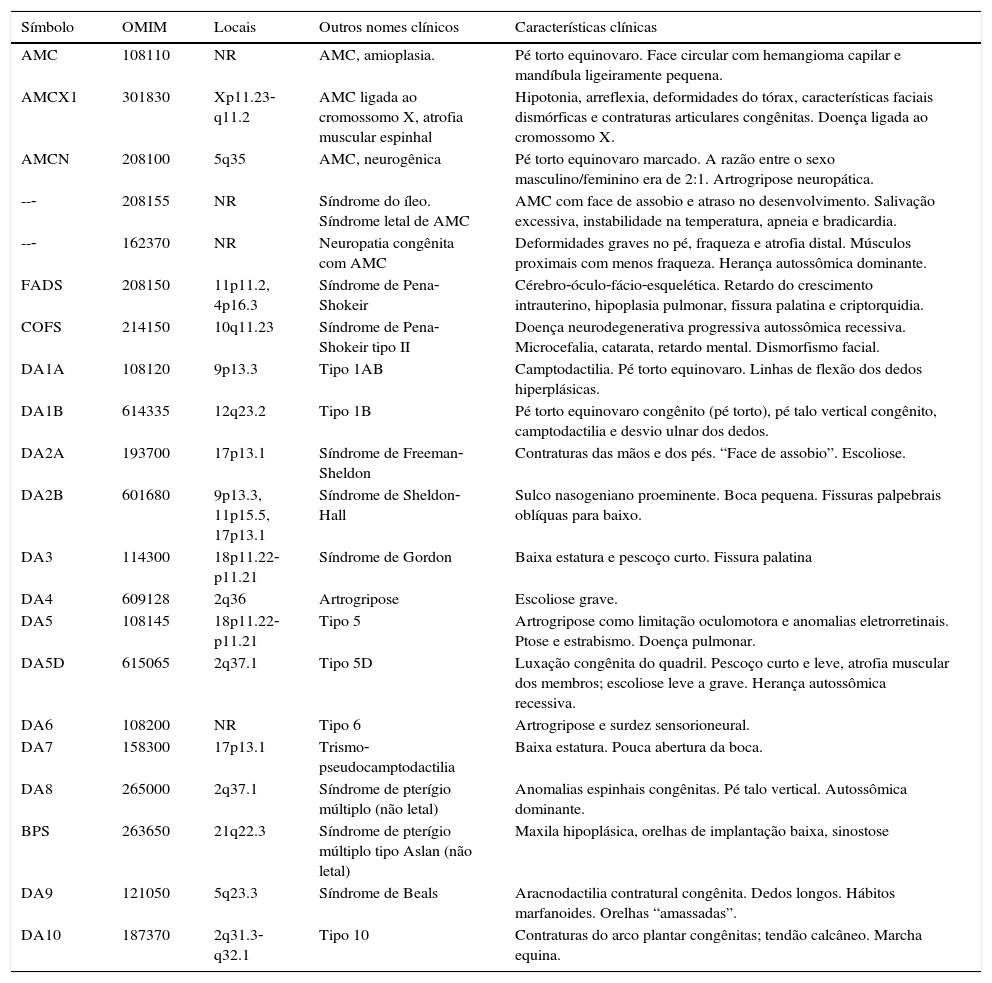
A tabela 2 inclui uma breve análise da literatura sobre diferentes variantes descritas de artrogripose múltipla congênita e distal. Em nossa série, há dois casos não familiares e não relacionados em que se observou consanguinidade. Além disso, também foram observados três casos familiares sem comprovação de consanguinidade (fig. 2).
Algumas variantes descritas na classificação da artrogripose múltipla congênita e distal
| Símbolo | OMIM | Locais | Outros nomes clínicos | Características clínicas |
|---|---|---|---|---|
| AMC | 108110 | NR | AMC, amioplasia. | Pé torto equinovaro. Face circular com hemangioma capilar e mandíbula ligeiramente pequena. |
| AMCX1 | 301830 | Xp11.23‐ q11.2 | AMC ligada ao cromossomo X, atrofia muscular espinhal | Hipotonia, arreflexia, deformidades do tórax, características faciais dismórficas e contraturas articulares congênitas. Doença ligada ao cromossomo X. |
| AMCN | 208100 | 5q35 | AMC, neurogênica | Pé torto equinovaro marcado. A razão entre o sexo masculino/feminino era de 2:1. Artrogripose neuropática. |
| --‐ | 208155 | NR | Síndrome do íleo. Síndrome letal de AMC | AMC com face de assobio e atraso no desenvolvimento. Salivação excessiva, instabilidade na temperatura, apneia e bradicardia. |
| --‐ | 162370 | NR | Neuropatia congênita com AMC | Deformidades graves no pé, fraqueza e atrofia distal. Músculos proximais com menos fraqueza. Herança autossômica dominante. |
| FADS | 208150 | 11p11.2, 4p16.3 | Síndrome de Pena‐Shokeir | Cérebro‐óculo‐fácio‐esquelética. Retardo do crescimento intrauterino, hipoplasia pulmonar, fissura palatina e criptorquidia. |
| COFS | 214150 | 10q11.23 | Síndrome de Pena‐Shokeir tipo II | Doença neurodegenerativa progressiva autossômica recessiva. Microcefalia, catarata, retardo mental. Dismorfismo facial. |
| DA1A | 108120 | 9p13.3 | Tipo 1AB | Camptodactilia. Pé torto equinovaro. Linhas de flexão dos dedos hiperplásicas. |
| DA1B | 614335 | 12q23.2 | Tipo 1B | Pé torto equinovaro congênito (pé torto), pé talo vertical congênito, camptodactilia e desvio ulnar dos dedos. |
| DA2A | 193700 | 17p13.1 | Síndrome de Freeman‐Sheldon | Contraturas das mãos e dos pés. “Face de assobio”. Escoliose. |
| DA2B | 601680 | 9p13.3, 11p15.5, 17p13.1 | Síndrome de Sheldon‐Hall | Sulco nasogeniano proeminente. Boca pequena. Fissuras palpebrais oblíquas para baixo. |
| DA3 | 114300 | 18p11.22‐p11.21 | Síndrome de Gordon | Baixa estatura e pescoço curto. Fissura palatina |
| DA4 | 609128 | 2q36 | Artrogripose | Escoliose grave. |
| DA5 | 108145 | 18p11.22‐p11.21 | Tipo 5 | Artrogripose como limitação oculomotora e anomalias eletrorretinais. Ptose e estrabismo. Doença pulmonar. |
| DA5D | 615065 | 2q37.1 | Tipo 5D | Luxação congênita do quadril. Pescoço curto e leve, atrofia muscular dos membros; escoliose leve a grave. Herança autossômica recessiva. |
| DA6 | 108200 | NR | Tipo 6 | Artrogripose e surdez sensorioneural. |
| DA7 | 158300 | 17p13.1 | Trismo‐pseudocamptodactilia | Baixa estatura. Pouca abertura da boca. |
| DA8 | 265000 | 2q37.1 | Síndrome de pterígio múltiplo (não letal) | Anomalias espinhais congênitas. Pé talo vertical. Autossômica dominante. |
| BPS | 263650 | 21q22.3 | Síndrome de pterígio múltiplo tipo Aslan (não letal) | Maxila hipoplásica, orelhas de implantação baixa, sinostose |
| DA9 | 121050 | 5q23.3 | Síndrome de Beals | Aracnodactilia contratural congênita. Dedos longos. Hábitos marfanoides. Orelhas “amassadas”. |
| DA10 | 187370 | 2q31.3‐q32.1 | Tipo 10 | Contraturas do arco plantar congênitas; tendão calcâneo. Marcha equina. |
AMC, artrogripose múltipla congênita; DA, artrogripose distal; FADS, Sequência de deformação acinesia fetal; COFS, Síndrome cérebro‐óculo‐fácio‐esquelética; BPS, Síndrome de Bartsocas‐Papas; OMIM, Mendelian Inheritance in Man On‐Line; NR, não relatado.
 com o mesmo tipo de artrogripose múltipla congênita. As setas indicam os pacientes estudados. Os quadrados representam o sexo masculino e os círculos, o sexo feminino; os quadrados/círculos pretos representam os indivíduos afetados.")
Na AMC, uma contratura é a limitação de mobilidade em uma articulação específica; a AMC não representa um problema intrínseco da articulação durante o desenvolvimento fetal porque a organização das articulações parece normal. É a falta de mobilidade articular durante o crescimento fetal que está associada ao desenvolvimento de tecido conjuntivo extra ao redor das articulações. Essas deformidades normalmente são simétricas, não progressivas e envolvem mais de uma área do corpo.3 A AMC é muito evidente no nascimento e o diagnóstico é possível no pré‐natal com ultrassom em tempo real.16,17
Na AMC, os quatro membros estão envolvidos em 50‐60% dos casos, os membros inferiores em 30‐40% e em 10‐15% as contraturas são limitadas aos membros superiores.7 Em nossos casos, 54% mostraram envolvimento de todas as extremidades, 36%, dos membros inferiores, ao passo que os membros superiores estavam envolvidos em 10% e não foi observada diferença entre meninos e meninas. Esses dados são compatíveis com aqueles relatados na literatura internacional (fig. 1).3,7,13,14,18
Pelo menos 150 entidades clínicas específicas foram descritas com AMC. É evidente que múltiplos fatores etiológicos convergem para uma resposta comum, o que resulta em perda do movimento das articulações. A AMC também é conhecida por ocorrer como componente de um grande grupo de disfunções heterogêneas miopáticas, neurogênicas, vasculares e de outras anomalias, como displasia esquelética.3,7 A tabela 2 descreve um resumo atualizado das diferentes variantes descritas na classificação da artrogripose múltipla congênita e distal. O tecido conjuntivo anormal, especificamente o colágeno, pode interferir no desenvolvimento normal do osso, da cartilagem e dos tendões, pode resultar em contraturas da articulação e fibrose. Nesta série, observamos um paciente com síndrome de Ehlers‐Danlos tipo 3 e um com osteogênese imperfeita tipo I (pacientes 4 e 9, tabela 1).
O histórico de pré‐natal é muito importante no estudo da AMC; nesta série, foram observadas doenças maternais, como diabetes mellitus, infecção do trato urinário com febre e eclâmpsia. Houve também um evento de sangramento vaginal durante o primeiro trimestre de gravidez. A identificação desses fatores ambientais e não genéticos eventuais é importante para estabelecer tipos específicos de AMC. Nesses casos, existe a possibilidade de fatores ambientais terem contribuído para a etiologia da AMC.
Foi descrito que posições anormais do feto, gestação múltipla e anomalias estruturais do útero podem gerar limitação de movimentos nas articulações e, consequentemente, promover a AMC. Metade de nossos pacientes teve apresentação cefálica, ao passo que a outra metade teve uma das apresentações de risco consideradas (pélvica, transversal ou outra posição). Também observamos um nascimento múltiplo e um caso com anomalias no útero que também são consideradas fatores de risco.
Os modos específicos de herança da artrogripose são descritos com relação a formas específicas de contraturas. Às vezes a AMC é causada por um defeito em um único gene; nesses casos, é possível ter herança autossômica dominante, autossômica recessiva e ligada ao cromossomo X. No momento, foram relatados diversos loci relacionados a diferentes tipos de artrogripose, por exemplo: aracnodactilia contratural (5q23‐31), displasia diastrófica (5q31), atrofia muscular espinhal letal (5q13.3), artrogripose distal tipo I (9q21.2), sinfalangismo (9q), artrogripose letal ligada ao cromossomo X (Xp11.3‐q11.2) e oftalmoplegia (12p11.2‐q12) (tabela 2).10‐13,19‐24 Existem até alguns relatos de que um paciente com AMC tem uma duplicação de um cromossomo e a deleção de outro cromossomo.23
A análise familiar é uma parte importante da avaliação de uma criança com AMC; ela é necessária para sugerir o modo de herança e fornecer o risco de recidiva para os pais. Figura 2A: Família com três irmãos afetados (mãos e pés), apesar de não haver dados de consanguinidade, o traço mais provável é de transmissão autossômica recessiva e o mosaicismo germinal é outra possibilidade que não pode ser descartada. Figura 2B: Família com dois irmãos afetados (mãos e pés), traços de herança autossômica recessiva ou ligada ao cromossomo X como modo de transmissão mais provável e mosaicismo germinal também deve ser considerado. Figura 2C: Família com três membros afetados em três gerações (mãos e pés afetados). Essa genealogia sugere fortemente uma herança autossômica dominante. O fato de o paciente II‐1 não ser afetado pode ser explicado em virtude da penetrância incompleta.
A AMC também pode ser causada por aberrações cromossômicas numéricas ou estruturais. Essas anomalias são particularmente frequentes em casos de AMC associada a retardo mental. A maioria dessas anomalias cromossômicas está presente como novos eventos; isso significa que o risco de recidiva em parentes é muito baixo.25,26 Neste estudo, as anomalias cromossômicas estiveram presentes em dois de 50 casos, um com 47,XY,+21 e outro com 47,XXY. Contudo, essas anomalias cromossômicas não estão associadas à AMC. Nenhum dos dois pacientes com retardo mental nesta série (2 e 8) apresentou anomalias cromossômicas, conforme mostrado pela análise do cariótipo feita em leucócitos do sangue periférico. Contudo, não podemos descartar completamente a possibilidade de um mosaicismo cromossômico de pequena proporção, pois nenhum outro tecido, como a pele (fibroblastos), foi usado na análise do cariótipo.25 Estudos cromossômicos em pacientes com AMC e retardo mental ou doenças multissistêmicas são muito importantes para o reconhecimento de síndromes clínicas com implicações cromossômicas.
O diagnóstico pós‐natal de AMC exige uma compilação de prontuários médicos, histórico familiar (ou seja, outros parentes afetados, hiperextensibilidade, hipotonia e outros sinais relacionados à doença), dados laboratoriais (enzimas séricas), análise radiográfica e estudos eletrofisiológicos e patológicos (incluindo biópsia muscular e do nervo). Para os casos considerados eventuais, em que nenhum histórico de saúde relevante é encontrado e nenhuma herança mendeliana padrão pode ser observada, o risco empírico de recidiva é de cerca de 3‐5%. O tratamento adequado na AMC deve ser multidisciplinar, com a participação de um pediatra especialista em ortopedia, um fisioterapeuta, um geneticista, um neurologista, dentre outros. O objetivo principal do tratamento é melhorar a função dos membros afetados. O manejo de complicações possivelmente evitáveis (luxação das articulações, osteoartrite e escoliose) é fundamental para garantir a qualidade de vida dos pacientes. Por outro lado, os programas de reabilitação facilitam e promovem a função independente nas atividades diárias. E por fim, conforme exposto acima, essa doença envolve fatores genéticos e ambientais. Portanto, o aconselhamento genético deve ser levado em consideração caso a família planeje ter outro filho.27,28
Neste trabalho, a classificação da AMC foi atualizada de acordo com a literatura. Descobrimos que existem apenas alguns relatos sobre AMC na população mexicana: em 1976, um relatório descreveu 83 casos,29 depois 46 em 199130 e um manuscrito relacionado a artrogripose distal tipo IIB em 1999.22 Neste estudo, incluímos 50 novos casos com grau variado de gravidade, alguns deles associados a outros problemas médicos, o que resultou em graves limitações nesses pacientes. Portanto, é importante estabelecer programas de fisioterapia e reabilitação específicos para os pacientes de acordo com as características específicas e a evolução de cada caso. Como em estudos anteriores, reiteramos que é necessária uma abordagem multidisciplinar com cuidado médico, cirúrgico, de reabilitação, social e psicológico, incluindo aconselhamento genético, para atingir uma gestão bem‐sucedida desses pacientes.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Valdés‐Flores M, Casas‐Avila L, Hernández‐Zamora E, Kofman S, Hidalgo-Bravo A. Characterization of a group unrelated patients with arthrogryposis multiplex congenita. J Pediatr (Rio J). 2016;92:58–64.




