

To evaluate, in a sample of patients with disorders of sex development (DSD), data related to the age at referral and their correlation with the initial complaints, gender at referral, defined gender after diagnosis and etiological diagnosis.
MethodsRetrospective review of the age at the first consultation and the reason for it, initial social gender and gender after the diagnosis, karyotype and etiological diagnosis of all cases treated at a DSD outpatient clinic between 1989 and 2016. Cases that did not involve DSD and DSD diagnoses that do not usually involve ambiguous genitalia, thus not requiring specialized monitoring, were excluded.
ResultsOf the 1793 treated cases, 1139 were diagnosed with some type of DSD. This study excluded 430 cases (272 with Turner's syndrome, 66 with Klinefelter syndrome, and 92 with pure gonadal dysgenesis), thus a total 709 individuals were included. Of these, 82.9% were referred due to ambiguous genitalia; only one-quarter were still in the first month of life, and 6.6% were referred due to pubertal delay, with most of them aged 10 years or older. Of these patients, 68.6% had a diagnosis of XY DSD, 22.4% of XX DSD, and 9% of sex chromosome abnormalities.
ConclusionsThis study presents the largest series in the literature of patients with DSD treated in a single center. The time of referral of the majority of patients with ambiguous genitalia fell short of the ideal, and milder cases of ambiguous genitalia and many with pubertal manifestations were referred even later. The results reinforce the importance of continuing education for professionals who will have the first contact with these patients, mainly pediatricians and neonatologists.
Avaliar em uma amostra de pacientes com distúrbios da diferenciação do sexo (DDS), dados relacionados à idade, ao encaminhamento e sua correlação com as queixas iniciais, ao sexo ao encaminhamento e ao sexo final e diagnóstico etiológico.
MétodosRevisão retrospectiva da idade por ocasião da primeira consulta e motivo da mesma, sexo social inicial e após definição do diagnóstico, cariótipo e diagnóstico etiológico de todos os casos atendidos em um ambulatório especializado em DDS entre 1989 e 2016. Foram excluídos casos que não compreendiam DDS e diagnósticos de DDS que não cursam comumente com ambiguidade genital, não necessitam de acompanhamento especializado.
ResultadosDos 1.793 casos atendidos, 1.139 foram diagnosticados com algum DDS. Excluíram-se 430 (272 síndrome de Turner, 66 síndrome de Klinefelter e 92 disgenesia gonadal pura), totalizando 709. Desses, 82,9% foram encaminhados por ambiguidade genital, somente um quarto ainda no primeiro mês de vida e 6,6% por atraso puberal, a maioria com 10 anos ou mais; 68,6% tiveram diagnóstico de DDS XY; 22,4% DDS XX e 9% de anomalias dos cromossomos sexuais.
ConclusõesEste estudo apresenta a maior casuística na literatura de pacientes com DDS atendidos em um único serviço. O momento de encaminhamento da maioria dos pacientes com ambiguidade genital foi aquém do ideal e casos mais leves de ambiguidade e muitos com manifestações puberais foram encaminhados ainda mais tardiamente. Os resultados reforçam a importância do ensino continuado a profissionais que terão o primeiro contato com esses pacientes, principalmente pediatras e neonatologistas.
Since the Chicago Consensus was published in 2006, Disorders of Sex Development (DSD) have been defined as congenital conditions in which the development of the chromosomal, gonadal, or anatomical sex is atypical.1 Thus, etiological conditions and diverse clinical manifestations, most of them rare, are encompassed.
Although these conditions result from predominantly congenital genetic defects, DSD does not always manifest at birth, and it is primarily up to the neonatologist, the general pediatrician, or even the clinician to consider this diagnosis.1–3
In cases of alterations already observed at birth, the child may have different degrees of ambiguous genitalia.1–4 During childhood or adolescence, DSD should be suspected in cases of delayed onset of puberty for age or in those with a progression different from what is expected.1,2,4–8 Finally, in adults, early gonadal failure and infertility are conditions that should lead to suspicion of DSD.1,2
Although rare, DSD do not characterize solely organic diseases, but also a social disability, due to the possible difficulty in defining the biological sex and the misunderstandings that can result from the unawareness of the differences regarding biological sex concepts, defined based on the child's sexual organs, gender identity, the psychological gender, with which the individual best identifies, and the sexual identity, the gender that represents the individual's desires and affections.9
Therefore, it is recommended that DSD cases should be treated at specialized centers by an experienced multidisciplinary team, prepared to deal not only with the diagnostic investigation and therapeutic conduct, but also with the psychological support required during the process.1,2
There are scarce data in the literature on the frequency of the diverse DSD etiologies in different age groups (newborns and infants, preschool and school-aged children, adolescents and adults), as well as about the initial clinical complaints, karyotype results, gender in which the individual was raised and final gender of these patients. Therefore, the objectives of this study were to evaluate, according to the age group, the etiology of the DSD cases treated at a specialized center, as well as the initial complaints, the karyotypes, the initial and final genders, and their relationships, considering the time of referral of these patients.
MethodsThe sample consisted of individuals with DSD referred to a specialized center between January 1989 and December 2016, at different age groups, who already had a confirmed etiological diagnosis at the time of the present study.
The data obtained from the patients’ medical records were as follows: age and social gender at the time of the first consultation, complaint at the time of referral to the service, karyotype, confirmed etiological diagnosis, and final gender, defined at the end of the etiological investigation. The main complaints were separated into ambiguous genitalia, pubertal delay, hypogonadism, gynecomastia, amenorrhea, infertility, dysmorphisms or malformations, and genetic counseling. The etiological diagnosis was confirmed according to the classification suggested by the Chicago Consensus.1
Cases that did not have a confirmed diagnosis of DSD and cases of Turner's syndrome, Klinefelter syndrome, and pure gonadal dysgenesis were excluded because, generally, they do not have ambiguous genitalia and can be diagnosed and evaluated at endocrinological services not specialized in DSD.
The study was approved by the Research Ethics Committee (CAAE 97392018.0.0000.5404).
The statistical analysis was performed using the software SPSS (IBM SPSS Statistics for Windows, Version 20.0. NY, USA). The data are presented in tables with absolute and relative frequency of each reason for the consultation and the etiological diagnosis, divided into the different age groups: 0–1 months, 2–12 months, 13–119 months, 120–239 months, and 240 months and older.
ResultsAll patients treated at the service between January 1989 and December 2016 had their data obtained and analyzed, totaling 1793 individuals. Of these, 1139 cases had a confirmed final diagnosis of a DSD etiology. The 654 individuals whose final diagnosis was not DSD included cases of isolated congenital genital malformation (42 cases), multiple congenital malformations including the genitalia (17 cases), patients with signs suggestive of Turner's syndrome or Klinefelter syndrome, but without karyotype alterations (392 and 142 cases, respectively), pubertal delay not related to DSD (29 cases), and transgender individuals (32 cases). These cases were excluded from the analysis and were considered errors of referral to the outpatient clinic (36.4%).
Another 430 patients, those with a diagnosis of Turner's syndrome 272 (23.8%), Klinefelter syndrome 66 (5.7%), or pure gonadal dysgenesis 92 (8%), were also excluded. Therefore, 709 cases were included.
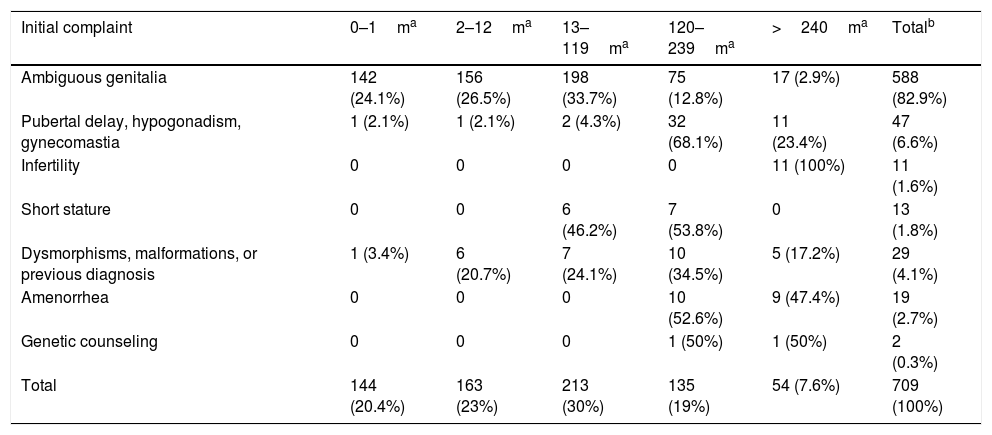
Table 1 shows the reasons for referral according to the age group. There was a predominance of cases with ambiguous genitalia 588 cases (82.9%), of whom 50.6% were referred in the first 12 months of life. Of the 47 cases of pubertal delay, hypogonadism, or gynecomastia, 91.5% (43 cases) were referred after 10 years of age. Patients referred at less than 1 year of age were cases of early diagnoses of hypogonadism at mini-puberty, one with pan-hypopituitarism and the other with micro-orchidism. The 19 cases (2.7%) of amenorrhea were also referred after 10 years of age. The two cases referred for genetic counseling were pregnant women with a history of previous pregnancy of children with congenital adrenal hyperplasia.
Frequencies of the initial complaint by age group, in months, of the 709 cases of Disorders of Sex Development (DSD).
| Initial complaint | 0–1ma | 2–12ma | 13–119ma | 120–239ma | >240ma | Totalb |
|---|---|---|---|---|---|---|
| Ambiguous genitalia | 142 (24.1%) | 156 (26.5%) | 198 (33.7%) | 75 (12.8%) | 17 (2.9%) | 588 (82.9%) |
| Pubertal delay, hypogonadism, gynecomastia | 1 (2.1%) | 1 (2.1%) | 2 (4.3%) | 32 (68.1%) | 11 (23.4%) | 47 (6.6%) |
| Infertility | 0 | 0 | 0 | 0 | 11 (100%) | 11 (1.6%) |
| Short stature | 0 | 0 | 6 (46.2%) | 7 (53.8%) | 0 | 13 (1.8%) |
| Dysmorphisms, malformations, or previous diagnosis | 1 (3.4%) | 6 (20.7%) | 7 (24.1%) | 10 (34.5%) | 5 (17.2%) | 29 (4.1%) |
| Amenorrhea | 0 | 0 | 0 | 10 (52.6%) | 9 (47.4%) | 19 (2.7%) |
| Genetic counseling | 0 | 0 | 0 | 1 (50%) | 1 (50%) | 2 (0.3%) |
| Total | 144 (20.4%) | 163 (23%) | 213 (30%) | 135 (19%) | 54 (7.6%) | 709 (100%) |
Regarding the patients’ karyotypes, individuals with the 46,XY karyotype represented three times (68.6%) the number of 46,XX individuals (22.4%). Sexual chromosome abnormalities, characterized by structural or numerical alterations after ruling out Turner's and Klinefelter syndromes, comprised 9% of the cases.
Table 2 shows the frequencies of the initial and final genders and their relationships. Only 130 cases (18.3%) came to the service with an indefinite social gender. The patients were aged up to 12 months, of whom 99 (76.2%) were referred in the first month of life. Thirty cases (12%) required gender reassignment, 17 from male to female and 13 from female to male. In the end, there were 65% males and 35% females.
Frequencies of the initial gender, gender at referral, and final gender of the 709 cases of Disorders of Sex Development (DSD), defined after etiological diagnosis.
| Initial gender | Male final gender | Female final gender | Total |
|---|---|---|---|
| Undefined | 72 (55%) | 58 (45%) | 130 |
| Male | 376 (95%) | 17 (5%) | 393 |
| Female | 13 (7%) | 173 (93%) | 186 |
| Total | 461 (65%) | 248 (35%) | 709 |
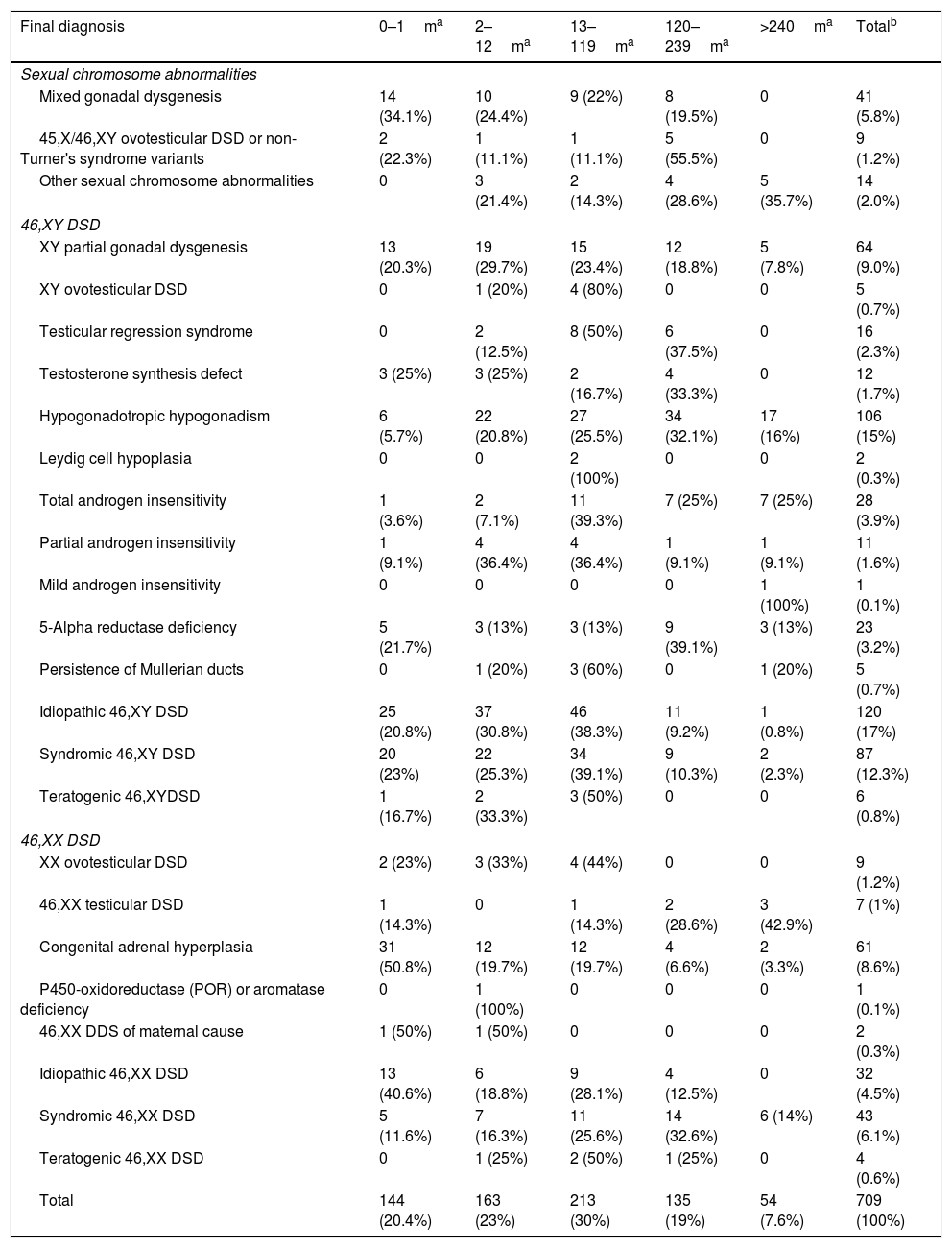
Regarding the diagnoses performed, Table 3 shows their distribution by age group. It was observed that more than half, 486 patients (68.6%), had a diagnosis of 46,XY DSD, 159 patients (22.4%) had a diagnosis of 46,XX DSD and 64 (9%) had sexual chromosome alterations. Among the cases of sexual chromosome abnormalities, the diagnoses of mixed gonadal dysgenesis predominated before 10 years of age, with the median age at the diagnosis of 4 months (CI: 0–204), while those with ovotesticular DSD had a similar distribution before and after 10 years of age, with a median age of 120 months (CI: 0–228). As for the other sexual chromosome abnormalities, diagnosis predominantly occurred after 10 years of age, with a median of 210 months (CI: 2–408). Regarding the 46,XY DSD cases, XY partial gonadal dysgenesis, testicular regression syndrome and ovotesticular XY DSD, both idiopathic and syndromic, predominatedup to 10 years of age, with the following respective medians and Confidence Intervals: 13.5 months (CI: 0–324), 82 months (CI: 2–216), 18 months (CI: 6–300) and 14 months (CI: 0–420). The diagnosis of total androgen insensitivity was greater after 1 year of age, with a median of 113 months (CI: 1–432) and the others were similar in all age groups. Finally, regarding the cases of 46,XX DSD, patients with congenital adrenal hyperplasia had a predominance of diagnosis up to one year of age, most within the first month of life, with a median of 1 month (CI: 0–432). The other diagnoses showed a similar distribution in the assessed age groups.
Frequencies of the different disorders of sex development (DSD) diagnoses by age group, in months, of the 709 cases.
| Final diagnosis | 0–1ma | 2–12ma | 13–119ma | 120–239ma | >240ma | Totalb |
|---|---|---|---|---|---|---|
| Sexual chromosome abnormalities | ||||||
| Mixed gonadal dysgenesis | 14 (34.1%) | 10 (24.4%) | 9 (22%) | 8 (19.5%) | 0 | 41 (5.8%) |
| 45,X/46,XY ovotesticular DSD or non-Turner's syndrome variants | 2 (22.3%) | 1 (11.1%) | 1 (11.1%) | 5 (55.5%) | 0 | 9 (1.2%) |
| Other sexual chromosome abnormalities | 0 | 3 (21.4%) | 2 (14.3%) | 4 (28.6%) | 5 (35.7%) | 14 (2.0%) |
| 46,XY DSD | ||||||
| XY partial gonadal dysgenesis | 13 (20.3%) | 19 (29.7%) | 15 (23.4%) | 12 (18.8%) | 5 (7.8%) | 64 (9.0%) |
| XY ovotesticular DSD | 0 | 1 (20%) | 4 (80%) | 0 | 0 | 5 (0.7%) |
| Testicular regression syndrome | 0 | 2 (12.5%) | 8 (50%) | 6 (37.5%) | 0 | 16 (2.3%) |
| Testosterone synthesis defect | 3 (25%) | 3 (25%) | 2 (16.7%) | 4 (33.3%) | 0 | 12 (1.7%) |
| Hypogonadotropic hypogonadism | 6 (5.7%) | 22 (20.8%) | 27 (25.5%) | 34 (32.1%) | 17 (16%) | 106 (15%) |
| Leydig cell hypoplasia | 0 | 0 | 2 (100%) | 0 | 0 | 2 (0.3%) |
| Total androgen insensitivity | 1 (3.6%) | 2 (7.1%) | 11 (39.3%) | 7 (25%) | 7 (25%) | 28 (3.9%) |
| Partial androgen insensitivity | 1 (9.1%) | 4 (36.4%) | 4 (36.4%) | 1 (9.1%) | 1 (9.1%) | 11 (1.6%) |
| Mild androgen insensitivity | 0 | 0 | 0 | 0 | 1 (100%) | 1 (0.1%) |
| 5-Alpha reductase deficiency | 5 (21.7%) | 3 (13%) | 3 (13%) | 9 (39.1%) | 3 (13%) | 23 (3.2%) |
| Persistence of Mullerian ducts | 0 | 1 (20%) | 3 (60%) | 0 | 1 (20%) | 5 (0.7%) |
| Idiopathic 46,XY DSD | 25 (20.8%) | 37 (30.8%) | 46 (38.3%) | 11 (9.2%) | 1 (0.8%) | 120 (17%) |
| Syndromic 46,XY DSD | 20 (23%) | 22 (25.3%) | 34 (39.1%) | 9 (10.3%) | 2 (2.3%) | 87 (12.3%) |
| Teratogenic 46,XYDSD | 1 (16.7%) | 2 (33.3%) | 3 (50%) | 0 | 0 | 6 (0.8%) |
| 46,XX DSD | ||||||
| XX ovotesticular DSD | 2 (23%) | 3 (33%) | 4 (44%) | 0 | 0 | 9 (1.2%) |
| 46,XX testicular DSD | 1 (14.3%) | 0 | 1 (14.3%) | 2 (28.6%) | 3 (42.9%) | 7 (1%) |
| Congenital adrenal hyperplasia | 31 (50.8%) | 12 (19.7%) | 12 (19.7%) | 4 (6.6%) | 2 (3.3%) | 61 (8.6%) |
| P450-oxidoreductase (POR) or aromatase deficiency | 0 | 1 (100%) | 0 | 0 | 0 | 1 (0.1%) |
| 46,XX DDS of maternal cause | 1 (50%) | 1 (50%) | 0 | 0 | 0 | 2 (0.3%) |
| Idiopathic 46,XX DSD | 13 (40.6%) | 6 (18.8%) | 9 (28.1%) | 4 (12.5%) | 0 | 32 (4.5%) |
| Syndromic 46,XX DSD | 5 (11.6%) | 7 (16.3%) | 11 (25.6%) | 14 (32.6%) | 6 (14%) | 43 (6.1%) |
| Teratogenic 46,XX DSD | 0 | 1 (25%) | 2 (50%) | 1 (25%) | 0 | 4 (0.6%) |
| Total | 144 (20.4%) | 163 (23%) | 213 (30%) | 135 (19%) | 54 (7.6%) | 709 (100%) |
The median of the DSD group as a whole was 24 months (0–696), whereas it was 12 months in the 591 cases of ambiguous genitalia (0–696).
DiscussionTo date, this study has described the largest worldwide cohort of patients from a single center, covering a 28 year period, with a confirmed diagnosis of one type of DSD, totaling 1139 cases. This was only possible because this is a national reference center dedicated to the integral, high-quality care of patients with DSD.
Similar samples verified in a literature review are from developing countries such as Brazil: in South Africa,10 346 patients were evaluated over 20 years; Thailand,11 117 patients over 20 years; Turkey,12 95 patients over 3 years; Indonesia,13 286 patients over 7 years; India,14 194 patients over 20 years; plus one developed country, Finland,15 with 550 patients over 10 years and two European multicenter studies, the I-DSD Registry,16 which is creating a database on DSD with 14 different countries and 649 patients, and the DSD Life,17 a consortium of 16 different hospital centers, which evaluated the quality of life of 1040 patients older than 16 years with DSD.
Although it is possible to claim that by suspecting DSD in so many patients the primary health care services reduce the risk of loss of diagnosis in some cases and that some tests, such as karyotyping, are not easily accessible outside specialized centers, the fact that more than one-third of referrals have been characterized as referral errors reinforces the need for training programs and the availability of didactic and support materials, so that non-specialist physicians, such as neonatologists, pediatricians, and clinicians, are able to refer only those patients with a real indication to the tertiary service, without overtaxing the health system and without causing unnecessary uncertainties and concerns to the families.4,18
It was also decided to exclude the cases diagnosed with Turner's syndrome, Klinefelter syndrome, or pure gonadal dysgenesis from the analysis by age group, although they are among the most common diagnoses, both in the present study's sample and in other cohorts; regarding the worldwide prevalence,5,8 these patients normally do not have ambiguous genitalia and do not need to be followed at multidisciplinary centers specialized in DSD. Therefore, those patients do not add value to the final objective of analysis regarding the referral process to the specialized center. The study carried out in India was the only one that also chose this exclusion, for the same reason.14
Patients with an initial complaint of genital ambiguity were referred in all age groups; however, only patients up to 12 months of age had no gender record, totaling 130 patients, which corresponds to less than half of the 298 patients with ambiguous genitalia referred in this age group.
Although the lack of gender assignment at birth leads to family distress and bureaucratic obstacles, since this is still a strong social factor, ideally it should be done only after adequate study of the case and after discussing it with the family members, considering the diagnosis, the surgical possibilities, the prospects for hormonal replacement therapy, and fertility, so that there will be a greater chance that the defined gender will be compatible with the individual's gender identity (which will only be defined later during the individual's life), so that it is not necessary to go through the process of gender reassignment, either in childhood or afterwards.18
Most patients with an undefined gender, 76.2%, were referred in the first month of life, a positive aspect to be highlighted that is of great value to the families, due to the prospect of an earlier completion of the investigation process and definition of the social gender. Among the patients with a defined gender at the referral, 55.4% were males and 26.2% were females.
Regarding the final gender assignment, 65% of the patients were male gender and 35%, female gender. Gender reassignment was performed only in individuals with ambiguous genitalia, occurring in 17 patients with an initial male gender registration (5%) and 13 initially registered as females (7%). In the Turkish sample, 39% of the patients had undefined gender at the referral, of whom 23.1% were males and 37.9% were females, and in the end, 44.3% were male gender and 55.7%, female gender.12
The main reason for referral in this study was the ambiguous genitalia in 82.9% of the patients. Although most were referred in the first year of life, this scenario is still far from the ideal, in which all cases with ambiguous genitalia would be detected in the maternity ward and the referral would be carried out at hospital discharge. Patients with ambiguous genitalia who were only referred after the age of 20, the worst-case scenario, had the most diverse diagnoses, mostly related to milder degrees of ambiguous genitalia and therefore identified later, such as XY partial gonadal dysgenesis, hypogonadotropic hypogonadism, total androgen insensitivity, 5α-reductase deficiency, and persistent Mullerian duct syndrome.
As expected, the majority of patients with pubertal delay, hypogonadism, and/or amenorrhea were referred between 10 and 19 years of age. However, there were also late referrals after 20 years of age in 23.4% of the patients with pubertal delay/hypogonadism and in 47.4% of those with amenorrhea, a fact that delayed the start of hormone replacement, causing growth impairment, bone mass reduction and, consequently, quality of life impairment in these patients.
Other analyzed samples do not separate the reasons for referral by age group, only their absolute frequency. In the study carried out in Thailand,11 36.8% of the patients were referred due to ambiguous genitalia, whereas in the Turkish study, 24.2% were referred due to the same cause12; these frequencies are similar to each other but lower than the frequency of the present study, at 82.9%. Pubertal delay was a reason for referral in 8.4% of patients in the Turkish study, similar to that of present study (6.6%), whereas amenorrhea was observed in 8.4%, higher than the percentage observed in this study (2.2%).
Regarding the diagnostic groups defined by the Chicago Consensus, in the present study 68.6% individuals were characterized as 46,XY DSD, 22.4% as 46,XX DSD and 9% as DSD due to sexual chromosome abnormalities. The cohorts from South Africa,10 Indonesia,13 India,14 and the I-DSD Registry16 showed similar proportions, with more than half of the cases representing 46,XY DSD, and sexual chromosome abnormalities representing a maximum of 10% of the cohort. The greater complexity of male sexual differentiation partially explains the existence of a higher number of cases of DSD with ambiguous genitalia in individuals with the 46,XY karyotype.19 In the studies carried out in Thailand,11 Turkey,12 and the DSD Life,17 there was a relative preponderance of sexual chromosome abnormalities (53%, 27.4%, and 54.3%, respectively) in relation to 46,XY DSD (17.1%, 47.3% and 21.4%, respectively). However, in the Finnish study,15 there were more cases of sexual chromosome abnormalities (37.1%) in comparison to the 46,XX DSD cases (9.6%). The reasons for that would be because these cohorts did not exclude individuals with Turner's syndrome and Klinefelter syndrome and in the Finnish study, due to the higher number of cases with a prenatal diagnosis of chromosomal abnormalities.
Finally, regarding the frequency of specific diagnoses and their age distribution, it is worth mentioning that among the 46,XY DSD group, 3.9% had a diagnosis of total androgen insensitivity, with a median age at diagnosis of 9.4 years. The proportion is similar to the group from Thailand,11 with a diagnosis rate of 5.1%, but in this case, the median age was 3.3 years, since many cases were diagnosed due to presence of an inguinal mass in childhood. In the study from India,14 the frequency was 1.5%, with a median age at diagnosis of 18 years. A Danish study20 that evaluated only cases of women with XY karyotype defined the median age at diagnosis as 7.5 years, also due to ambiguous genitalia or family history.
In XY partial gonadal dysgenesis, the frequency was 9% in this study, higher than in the South-African study10 and the DSD Life,17 at 1.1% and 3.5%, respectively, but similar to those from India14 and Thailand,11 at 5.6% and 6.0%, respectively. The median age at diagnosis of the present sample was 1.1 years, lower than that in the study carried out in Thailand, of 4.3 years.
Regarding testicular regression syndrome, the diagnostic frequency in the present study was 2.3%, with a median age of 6.8 years, well below the Finnish rate of 4.9%,15 with a median of 0.84 years.
Regarding the cases of 46,XX DSD, it is important to highlight the diagnosis of congenital adrenal hyperplasia. The percentage of 8.6% is low in relation to the worldwide rate21 and those of the majority of the studies: 26.5% in Thailand,11 16.8% in Turkey,12 13.9% in Indonesia,13 26.8% in India14 and 21.7% in the DSD Life.17 This is because in the present service there is another specific outpatient clinic for adrenal hyperplasia, which concentrates the patients diagnosed through neonatal screening and the referrals for late forms. The median age at diagnosis was 1 month, which demonstrates the efficacy of the neonatal screening system, being similar to the studies conducted in India and Finland. The South-African cohort10 had a prevalence similar to the present study's (9%), but this is due to the absence of diagnoses caused by the lack of neonatal screening in the country. The Finnish rate15 was also low, 5.6%, but this is explained by the low proportionality of 46,XX DSD in that cohort.
As for the sexual chromosome abnormalities, the prevalence of mixed gonadal dysgenesis is higher in the present study (5.8%) in relation to those of South Africa (1.1%),10 Turkey (3.1%),12 Indonesia (2.7%),13 and India (2.5%),14 whereas it is similar to the DSD Life,17 at 4.3%, and lower than that of Thailand,11 at 8.5%. The median age at diagnosis in the present study was low (0.33 years), lower than those in the studies performed in Indonesia and India (9.5 and 10 years, respectively).
Overall, the median age of the DSD group in this study was 24 months, whereas for the 591 cases of ambiguous genitalia, it was 12 months. Although relatively low, it remains inadequate, possibly reflecting in part the lack of identification of some cases at birth, the lack of knowledge on the investigation process by the pediatricians who evaluate the DSD cases, and the low availability of investigation centers in a country such as Brazil, with continental dimensions.
In conclusion, this study brings relevant contributions aimed at improving care for individuals with DSD, supported by the representativeness of its sample. The analysis of several aspects reinforces the importance of continuing education for professionals in charge of the initial care and referral of these patients to reference services, mainly pediatricians and neonatologists, allowing earlier and more efficient diagnostic and therapeutic interventions aimed at reducing complications, both in the physical and psychosocial aspects of these individuals, providing better prospects for adaptation and quality of life for people with DSD and their families.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: El Beck MS, Germano CW, Barros BA, Andrade JG, Guaragna-Filho G, Paula GB, et al. Why pediatricians need to know the disorders of sex development: experience of 709 cases in a specialized service. J Pediatr (Rio J). 2020;96:607–13.
Study conducted at Universidade Estadual de Campinas (Unicamp), Faculdade de Ciências Médicas (FCM), Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo (GIEDDS), Campinas, SP, Brazil.




