
Cystic fibrosis (CF) is a severe autosomal recessive disease that results from mutations in a gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein, a chloride channel. This study aims to characterize the clinical and genetic features of a cohort of pediatric people with CF (PwCF) in the center of Portugal and to determine which ones are candidates for the new drugs modulating the CFTR channel.
MethodsA review of the demographic, genetic and clinical characteristics of PwCF undergoing follow-up at a CF reference center was carried out.
ResultsTwenty-three PwCF (12 male), with a median age of 12 years, were followed up. All patients carry the F508del mutation in at least one allele. Fifteen PwCF were F508del-homozygous, median BMI z-score was -0.13, all are pancreatic insufficient and median FEV1 value was 78.1%. These PwCF are eligible for dual therapy (lumacaftor/tezacaftor+ivacaftor) and for triple therapy (tezacaftor+ivacaftor+elexacaftor). PwCF with 711 +1G->T (n = 2), 2184insA (n = 1) mutations and a novel mutation c.3321dup (n = 1) have minimal function mutation and patients with a residual function mutation: R334W (n = 3) and P5L (n = 1) have a less severe phenotype. All these patients, because they also carry F508del mutation, are elegible to triple therapy.
ConclusionsGenetic and molecular characterization of PwCF poses an important step not just for CF diagnosis and prognosis which is tightly correlated with the clinical phenotype, but also for the eligibility of CFTR modulator drugs.
Cystic fibrosis (CF) is a severe autosomal recessive inherited disease with a higher incidence in Caucasians.1 The disease results from mutations in a gene located on chromosome 7, which encodes the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein present in the apical membrane of epithelial cells that functions as a chloride channel.1 In Portugal, a prevalence of 0.27 per 10,000 inhabitants and an incidence of 1 per 7500 newborns has been estimated.2-4
Over 2,000 mutations have been reported, for which only a minority has disease liability established [CFTR2 mutations database]. One mutation - F508del - is the most common among People with cystic fibrosis (PwCF)5,6 with an estimated worldwide prevalence of ~80%. Mutations in the CFTR gene are grouped into seven functional classes for which the same therapeutic strategy applies: class I - premature stop codons (resulting in unstable, truncated or absent protein); class II - traffic (defects in protein maturation and transport to the membrane); class III - gating (defects in the regulation of the opening of the channel in the apical membrane); class IV - conductance (defects in ion flow through the channel); class V – reduced protein levels (often resulting from anomalies in splicing; class VI (accelerated turnover) and class VII – no protein (frameshift, indels or large deletions that result in total absence of mRNA).5,6
The diversity and heterogeneity in the presentation of the disease and its evolution are reflected in the existence of classic and non-classic forms of CF, with different degrees of clinical severity, but most noticeably the latter with variability in organ involvement and delayed presentation.5 These ‘atypical’ features are essentially due to the different molecular defects present in the CFTR gene that correspond to different degrees of CFTR dysfunction, namely, in the epithelia of the respiratory, pancreatic, intestinal, hepatobiliary systems, male genital apparatus, and sweat glands. However, there is also phenotypic variability among PwCF with the same genotype, raising the question that other mechanisms and factors may influence the evolution of the disease, such as modifier genes and environmental elements.5
CF is multisystemic and despite the great clinical variability in organ involvement, CF predominantly affects the airways leading to progressive lung disease (the major cause of morbidity and mortality), and the gastrointestinal tract, including pancreatic insufficiency (PI) and malnutrition.1,5 Despite the typical clinical manifestations of classical CF, additional tests are necessary to confirm CFTR dysfunction and thus a diagnosis of CF. The sweat test with high values of chloride concentration (> 60 mEq/L) confirms a definitive diagnosis of CF.3,7 In Portugal, as of October 2013, newborn screening (NBS) for CF has been carried out, initially as a pilot study and since 2018 integrated into the NBS National Programme. Subsequent identification of two CF-causing mutations establishes a diagnosis of CF.7
Classic CF treatment focuses on the symptoms to prevent disease progression. However, new modulator therapies which treat the molecular defects of CFTR are now in clinical practice, but they are not yet universally available.6,8 Until now, four oral CFTR modulators have been approved for PwCF by the European Medicines Agency (EMA): ivacaftor (Kalydeco®), lumacaftor/ivacaftor combined (Orkambi®) and tezacaftor/ivacaftor combined (Symkevi®) and the Highly effective CFTR modulators (HECM) tezacaftor+ivacaftor+elexacaftor combined (Kaftrio®).6
Ivacaftor is a CFTR potentiator, which increases the channel open probability (gating).6,9 Therefore, ivacaftor could be of benefit in PwCF who have class III/IV mutations, in which the CFTR channel is present at the plasma membrane (PM), but has gating or conductance defects, respectively.8,9 Clinical trials on ivacaftor showed an improvement of forced expiratory volume in one second (FEV1) of approximately 10% in PwCF with gating mutations. Tezacaftor and lumacaftor are both correctors, which promote correct folding of F508del-CFTR, enabling some mutant protein to be correctly transported to the PM.6,8,9 PwCF with two F508del mutations are candidates for these drugs, as this mutation interferes with normal protein folding and traffic, being marked for early destruction in the proteasome.9 However, studies showed that treatment with a corrective drug in monotherapy is not effective, and therefore the combination of correctors with a potentiator is required. In F508del-homozygous PwCF, results from clinical trials with the combination of ivacaftor with lumacaftor (Orkambi®) showed a modest increase of 2.6-3% in FEV1, with a 40% reduction in the frequency of exacerbations; while the combination of ivacaftor with tezacaftor (Symkevi®) revealed an increase in FEV1 by 4% and a reduction of 35% of exacerbations.9 In Europe, F508del-homozygous PwCF can start therapy with Orkambi® from the age of two or therapy with Symkevi® after the age of twelve. A new triple combination: elexacaftor + ivacaftor + tezacaftor (Kaftrio®) has recently been approved by EMA for treating PwCF with F508del mutation in at least one allele after the age of twelve. Clinical trials on this new therapy achieved the best results, with an increase in FEV1 of 14.3%.6 These treatment strategies alongside the improvement in symptomatology management are expected to contribute to the increase in the average life expectancy of PwCF, which is currently in the 40s in the United States.10
It is consequently of interest to study PwCF followed up at Hospital Pediátrico - Centro Hospitalar e Universitário de Coimbra (HP-CHUC), Portugal, so that they are classified in the different functional classes according to the mutations found, establish the genotype/phenotype correlation, and select possible candidates for CFTR modulator therapies.
The aim of this study is to characterize clinically and genetically pediatric PwCF in central Portugal and to determine which ones are candidates for Kalydeco®, Orkambi® or Kaftrio®.
MethodsThe present study was approved by the board of the Centro Hospitalar e Universitário de Coimbra after a favorable report by the Health Committee (Ref. CHUC-080-16).
A retrospective observational study was performed, using data obtained from the last appointment of the clinical files of PwCF followed in HP-CHUC in 2019 where the following variables are routinely registered: gender, current age, age at diagnosis, genotype, nutritional status, bone impairment, pulmonary function, microbiology, and pancreatic and hepatic function.
Frequencies and respective percentages were used to summarise the count data. Results were presented individually for PwCF with rarer mutations. For analysis that included more patients, results were summarised using median values and the range was used to measure dispersion.
The criteria used in the diagnosis were: clinical characteristics compatible with CF, positive sweat test and genetic study with the identification of two disease-causing mutations.
The sweat chloride test was considered positive if a conductivity test was is ≥ 85 mmol/L7 and/or the chloride concentration test ≥ 60 mmol/L. Currently, the recommended sweat test is the measurement of chloride concentration, however, the diagnosis of older PwCF was made before this method was available.
Mutations were identified for all PwCF, most as part of the diagnostic approach at the National Health Institute Dr. Ricardo Jorge. In the last years, in the first stage, the F508del mutation is tested by analyzing the Amplification-Refractory Mutation System. If the result excludes F508del homozygosity, the genetic study will be continued using the Elucigene ® CF-EU2v1 kit (that searches for the 50 most frequent mutations in Europe) and the Elucigene ® CF Iberian Panel kit (search for the 12 most frequent mutations in the Iberian Peninsula). In cases of positive chloride concentration test and only one or no CF, mutation identified, complete sequencing of the CFTR gene by next-generation sequencing is done.2
Nutritional status was assessed according to the z-score of weight, height, and body mass index (BMI). The nutritional status was assessed using the following classification: i) severe malnutrition z-score BMI <-3, ii) moderate malnutrition z-score -3 to - 2, iii) normal z-score from -2 to 2 and, iv) obesity z-score ≥2.11,12 Bone density in the lumbar spine was assessed using DEXA osteodensitometry (dual-energy X-ray absorptiometry). The first bone density assessment was done between eight and ten years of age and results from the last DEXA scan were considered. if osteodensitometry Z-score is below -2 it is considered to be significantly decreased.3
Lung function was defined based on the FEV1, considering a value of ≥80% as normal. Spirometry tests are only applied to PwCF from the age of six.3,13 Chronic airway infection refers to patients in whom airway samples were culture positive for the same bacterium in more than 50% of the samples obtained over the last 12 months (minimum 4 samples per year).14
The exocrine pancreatic function was assessed using fecal elastase levels considering exocrine insufficiency mild, i.e., pancreatic sufficient (PS) (between 100 and 200 µg/g) and severe, i.e., pancreatic insufficient, PI (< 100 µg/g).7
Liver disease associated with CF is defined as the presence of at least two of the following: hepatomegaly, changes in transaminases or ultrasound-suggested anomalies.7
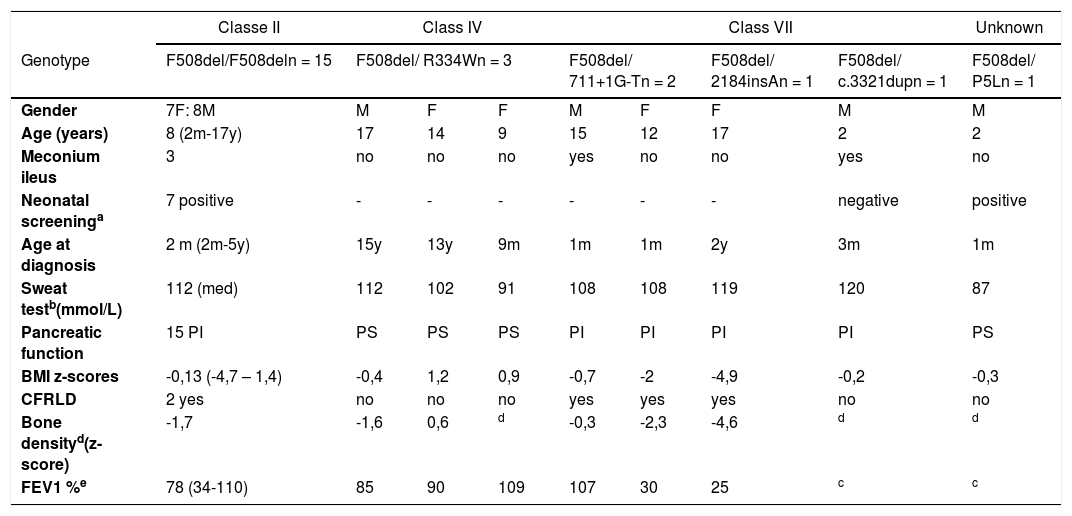
ResultsTwenty-three people from the center of Portugal with a diagnosis of CF were followed up at the HP-CHUC, and their CFTR genotypes were grouped according to the functional classes. Twelve were male and 11 female. The median age was 12 years (range: 2 months -18 years).
The initial manifestation that led to the CF diagnosis was: a positive NBS test (n = 7, 28%); respiratory symptoms and failure to thrive (n = 4, 16%); respiratory symptoms only (n = 5, 20%); failure to thrive only (n = 1, 4%); meconium ileus (n = 6, 24%); nasal polyps (n = 1, 4%); and hyponatremic and hypochloremic dehydration (n = 1, 4%).
The age of onset of clinical manifestations of the disease varied from the first days of life to 15 years, with a median of 3 months (Table 1).
Demographic and clinical characterization of PwCF according to functional classes of their genotype.
| Classe II | Class IV | Class VII | Unknown | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | F508del/F508deln = 15 | F508del/ R334Wn = 3 | F508del/ 711+1G-Tn = 2 | F508del/ 2184insAn = 1 | F508del/ c.3321dupn = 1 | F508del/ P5Ln = 1 | |||
| Gender | 7F: 8M | M | F | F | M | F | F | M | M |
| Age (years) | 8 (2m-17y) | 17 | 14 | 9 | 15 | 12 | 17 | 2 | 2 |
| Meconium ileus | 3 | no | no | no | yes | no | no | yes | no |
| Neonatal screeninga | 7 positive | - | - | - | - | - | - | negative | positive |
| Age at diagnosis | 2 m (2m-5y) | 15y | 13y | 9m | 1m | 1m | 2y | 3m | 1m |
| Sweat testb(mmol/L) | 112 (med) | 112 | 102 | 91 | 108 | 108 | 119 | 120 | 87 |
| Pancreatic function | 15 PI | PS | PS | PS | PI | PI | PI | PI | PS |
| BMI z-scores | -0,13 (-4,7 – 1,4) | -0,4 | 1,2 | 0,9 | -0,7 | -2 | -4,9 | -0,2 | -0,3 |
| CFRLD | 2 yes | no | no | no | yes | yes | yes | no | no |
| Bone densityd(z-score) | -1,7 | -1,6 | 0,6 | d | -0,3 | -2,3 | -4,6 | d | d |
| FEV1 %e | 78 (34-110) | 85 | 90 | 109 | 107 | 30 | 25 | c | c |
N, normal; PS, pancreatic sufficient; PI, pancreatic insufficient; y, years; m, months; M, male; F, female; BMI, body mass index; CFRLD-CF, related liver disease; FEV1, forced expiratory volume in 1 second.
Currently, 20% of PwCF have severe or moderate malnutrition and median BMI z-score is +0,25; 83% have exocrine pancreatic insufficiency, 16% have low bone mineral density (BMD) for chronological age (Table 1), 36% have lung function impairment and median FEV1 is 85%, 52% has chronic infection with one or more bacteria.
The genetic study identified six different mutations and, as expected, F508del was the most frequent one (allele frequency: 80%) present in homozygosity in 15 PwCF (60%).
Analyzing the data by functional classes, the authors found that the 15 PwCF with the homozygous F508del mutation (Class II) have a median age of eight years (2 months - 17 years); the median age at diagnosis is two months (ranging from 1 month to 5 years); they have a median BMI z-score of -0.13, ranging from -4.17 to +1.42; all these PwCF suffer from pancreatic insufficiency. The median sweat test value was 112 mmol/L (78 mmol/L to 124 mmol/L). The median FEV1 value was 78% (34% to 110%). Three (20%) of these PwCF had chronic lung infection with a bacterium, the most frequent being Staphylococcus aureus. Two have liver disease and two have a low bone density (Table 1).
The heterozygous PwCF all have the F508del mutation in one allele and in the other, mutations were: Class IV: R334W (n = 3); Class VII: 711 +1G->T (n = 2) and 2184insA (n = 1); not classified: P5L (n = 1). A new mutation - c.3321dup - was also not yet classified but it leads to a frameshift and therefore could be considered Class VII (n = 1). Functional studies to assess CFTR are in progress to confirm the classification of this mutation.
PwCF with class VII mutations are 2 to 18 years old, the age at diagnosis ranges from 1 month to 2 years old and all are PI. One of these patients has severe malnutrition and another one has moderate malnutrition. The median sweat test value was 114 mmol/L (range: 106-123 mmol/L). The median FEV1 value was 68% (range: 22-107%). In this group, all patients have chronic infection with a bacterium, the most frequent being Pseudomonas aeruginosa and Burkholderia spp. Three have liver disease and two suffer from low bone density. The new mutation was found in a 2-year-old male toddler with a clinical history highly suggestive of CF (meconium ileus, PI, chronic lung infections, and respiratory failure) and, although his NBS test was negative, he had two positive sweat chloride tests.
Class IV PwCF is represented here by three patients who are 9, 14 and 17 years old. Two of these PwCF were diagnosed as adolescents. They all are PS, they have an adequate nutritional status and their pulmonary function is normal. Two PwCF have a chronic lung infection, one with Pseudomonas aeruginosa and the other one with Staphylococcus aureus.
The child with the unclassified P5L mutation is 2 years old, had an adequate nutritional status, PS and with no respiratory symptoms.
Two homozygous PwCF for the F508del mutation were already in combination therapy lumacaftor plus ivacaftor. One 16-year old girl had a severely compromised lung function (40% predicted FEV1 and chronic lung infection with Burkholderia cepacia), PI and moderate undernutrition when treatment was started. After two years in treatment, her nutritional status improved and is now adequate and her lung function stabilized. Another 13-year old girl who has been on therapy for almost one year remained with severely compromised lung function (FEV1 34%) but her nutritional status slightly improved is now adequate.
DiscussionThe aim of this study was to clinically and genetically characterize pediatric PwCF in central Portugal and to determine which ones are candidates for new CFTR modulators. The authors found that analyzing clinical data and grouping patients in functional classes according to their genotype gives us an idea of the severity and prognosis of the disease in each individual patient. It also helps to identify whose patients may benefit from available CFTR modulators for which indications are mutation-specific.
The authors are aware of the limitations of the present study as it is based on a small number of PwCF from only one center. Particularly, conclusions when comparing characteristics from different functional classes are limited because of the small number of PwCF in each class.
Comparing the current group of PwCF under follow-up at HP-CHUC with the pediatric population described in the CF Foundation 2018 annual report (US), there are no significant differences in terms of nutritional status (median BMI z-score -0.2 vs +0,2, respectively). Lung function is slightly decreased in the study's PwCF but still in the normal range (FEV1 82% vs 97%, respectively).10 The most frequent initial manifestations did not deviate from the classically referred in the literature, but the diagnosis was more challenging in some less common initial manifestations, such as nasal polyps and hyponatremic hypochloremic dehydration.15
The frequency of the F508del mutation was equal to the 80% reported for Portugal, as well as for Northern and Central European countries.16
There was great phenotypic variability among PwCF with the same genotype, particularly in PwCF homozygous for F508del or with class VII mutations, which appear to be independent of the age/duration of the disease. In the same group, there were PwCF with malnutrition and impaired lung function and others with adequate nutritional status and normal pulmonary function, which raises the question that genetic or environmental factors may also influence the severity of the disease. Further studies are in order to analyze other factors, like socioeconomic status or compliance to therapy.17
Interestingly, the patient with the new class VII mutation and meconium ileus as initial manifestation had a false negative NBS test. This fact, which is described in the literature, is justified by the earlier and severe destruction of the pancreas during gestation and the consequent lower concentration of IRT and PAP at birth.2,6
The authors found that the three PwCF with Class IV mutations seems to have a less severe phenotype. This may be explained by the residual function of CFTR protein present in patients with these mutations.1 Previous studies have also described significantly lower sweat chloride values in PwCF with class IV mutations in comparison to PwCF homozygous for F508del and a later age at diagnosis.18
Since the approval of Orkambi® and Symkevi® drugs for PwCF homozygous for F508del, the present study's 15 PwCF would benefit from it. However, within our patients only 2 PwCF are currently on Orkambi® therapy. Also in the CF Foundation 2018 report (US) about 20% of PwCF considered eligible for treatment with CFTR modulators are not taking it.10 In fact these drugs are not yet accessible to all PwCF in all European countries. Currently, great hope is being placed on the HECM therapy,19 it was licensed for use by PwCF aged over 12 years who carry at least one copy of the F508del mutation.6 All of the study's PwCF will be eligible for HECM triple therapy when they reach 12 years of age, including F508del-homozygous, F508del-heterozygous with a minimal function mutation (711 +1G->T, 2184insA and c.3321dup) and F508del-heterozygous with residual function mutation (R334W and P5L).
Because no PwCF carries a gating mutation, Kalydeco® would not be indicated for any of the studied patients.
Ideally, all patients that fulfill the approved indications should immediately benefit from treatment, particularly with HECM which has shown better results. However, negotiations between individual country governments and the pharmaceutical industry are underway but the high cost has been an issue. For now, centers must decide which patients should start these therapies first. This raises the question: should we choose PwCF that already have severe and most probably irreversible consequences of their disease or should we choose patients that still have normal lung, pancreatic or liver function before they start to deteriorate? Should we choose adults or children?
Clinical trials that led to CFTR modulators approval are based on clinical outcomes, the main one being FEV1 improvement, however, an important piece of data to take into account when analyzing the cost-effectiveness of these drugs is the fact that, although the improvements in lung function are modest, the simple fact that they are potentially able to stabilize the progression of the disease, particularly if administered early in life and before irreversible damage, can prove to be of great importance for patients, health professionals and families struggling to contain the inexorable clinical deterioration characteristic of CF.
In conclusion, the genetic and molecular characterization of PwCF poses an important step not just for CF diagnosis and prognosis which is tightly correlated with the clinical phenotype, but also for the eligibility of CFTR modulator drugs, namely HECM.




