
This article presents a clinical and cytogenomic approach that focuses on the diagnosis of syndromic oral clefts (OCs).
MethodsThe inclusion criteria were individuals with OC presenting four or more minor signs and no major defects (non-syndromic oral clefts [NSOCs]) as well as individuals with OC presenting at least another major defect, regardless of the number of minor signs (syndromic oral clefts [SOCs]). The exclusion criteria included NSOC with less than four minor signs, SOC with known etiology, as well as atypical oral clefts.
ResultsOf 1647 individuals with OC recorded in the Brazilian Database of Craniofacial Anomalies, 100 individuals were selected for chromosome microarray analysis (CMA). Among these, 44 individuals were clinically classified as NSOC and 56 as SOC. CMA was performed for both groups, and abnormal CMA was identified in 9%, all previously classified as SCO. The clinical and CMA data analyses showed a significant predominance of abnormal CMA in individuals classified as SOC (p = 0.0044); prematurity, weight, length, and head circumference at birth were significantly lower in the group with abnormal CMA. Besides, minor signs were significantly higher in this group (p = 0.0090).
ConclusionThe rigorous selection of cases indicates that the significant variables could help in early recognition of SOC. This study reinforces the importance of applying the CMA technique to establish the diagnosis of SOC. This is an important and universal issue in clinical practice for intervention, care, and genetic counseling.
The success of a treatment depends on several factors. With regard to oral clefts (OC), such congenital defects are curable if therapeutic measures are taken in a timely manner.1 With a prevalence of 1 in 500–2500 births, OCs are recognized as a public health problem not only due to their prevalence but also due to their morbidity, as well as the need for individualized, long-term, multi-professional teams and technology-dependent follow-up.1,2 The recognition of the etiological factors and the diagnosis are crucial to personalized treatment planning and the formulation of public health strategies.3
Approximately 70% of OCs are classified as non-syndromic (NSOC) and have a multifactorial inheritance, whereas the group of typical syndromic oral clefts (SOCs) comprises more than 500 Mendelian conditions, chromosomal abnormalities, embryopathies due to teratogens, and multiple congenital defects whose etiology remains unknown.1,4
Differentiating NSOCs from SOCs is an important issue within clinical practice, especially in newborns with no additional major external anomalies. In many situations, the phenotype is subtle in this age group, hindering the early diagnosis and subsequent specific therapeutic interventions. Minor defects are morphological abnormalities that do not imply significant functional and/or cosmetic impairments, while the major ones correspond to morphological changes that have significant health impacts and may lead to death, permanent disability, or severe aesthetic impairment.5–7
In 20% of newborns with three or more minor signs, a major sign is also detected.6–9 It is noteworthy that although minor signs have predictive value for identifying major signs,6–9 there is low concordance in their clinical description and importance, leading to discrepancies among various studies.6,7 Clinical follow-up favors the diagnostic suspicion of SOC, since minor anomalies are more frequent at ages between 4 and 6 years.5,10
Chromosome microarray analysis (CMA) was employed as an important diagnostic tool for patients with neuropsychomotor developmental delay/intellectual disability, autism spectrum disorders, or multiple congenital anomalies.11,12 However, the value of CMA in the diagnosis of SOC has been little explored. In fact, only ten articles reporting a series of OC using CMA were identified up to November 2019.4,13–21
CMA detects the copy number variations (CNVs), which could be causative of SOC. CNVs are structural genomic imbalances that contain duplications or deletions of a DNA fragment. Although rarely directly related to the clinical condition, they can change the copy number of a gene and contribute to the modulation of gene expression, playing an important role in the susceptibility to or protection from complex phenotypes.13 Furthermore, CNV screening has proven to be a powerful strategy in identifying candidate genes and/or chromosomal regions involved in OC genesis, effectively complementing traditional genetic mapping strategies.4,13–21
This article presents a clinical and cytogenomic approach that focuses on the diagnosis of SOC.
MethodsThis is a multicenter, cross-sectional, and descriptive study based on the primary data of 1647 individuals with OC in The Brazilian Database on Craniofacial Anomalies (BDCA)22 registered from September 2008 to September 2018. It was approved by the Unicamp Research Ethics Committee (CAAE 35316314.9.1001.5404). Patients were personally assessed by geneticists at eight collaborating centers of Brazil´s Craniofacial Project. Clinical data (anamnesis and physical examination) were standardly inserted in the BDCA through Cranflow, an application developed by the present team.22 Biological samples were sent to and stored in a biorepository located in Campinas (São Paulo). Individual results were mailed to the assistant geneticist to contribute to the genetic counseling of the families.
The inclusion criteria were individuals with OC presenting four or more minor signs and no additional major defects (NSOCs) and individuals with OC presenting at least another major defect regardless of the number of minor signs (SOCs). These criteria were based on previous studies about the predictive value of minor signs with regard to OC.6–9 CMA was performed in 100 cases of OC recorded in the BDCA, preserving the national comprehensiveness.
The exclusion criteria included NSOCs with less than four minor signs, SOCs with known etiology (chromosomal or monogenic), as well as atypical oral clefts and cases where biological samples were unavailable for CMA.
The CMA technique was performed using the CytoScan HD and CytoScan 750 K arrays (Affymetrix® – Santa Clara, CA, United States), according to the manufacturer’s recommendations. The CNVs were analyzed using the Chromosome Analysis Suite v.3.3 (ChAS) software. The number of markers was set to a minimum of 25 for deletion and 50 for duplication. For mosaic alterations, a minimum number of 5000 markers was selected, for deletions as well as duplications. Regions of homozygosity (ROH) were investigated by selecting 500 markers as the minimum and 1500 Kb as the minimum size. Thresholds were determined according to company recommendations (Affymetrix® – Santa Clara, CA, United States) to be reliable for the detection of CNVs.
The interpretation of the chromosomal imbalances was based on international recommendations, according to the protocol developed by the FCM/UnicampCytogenetics and Cytogenomics Laboratory.23,24 All CNVs were compared with data derived from the international databases: the Database of Genomic Variants (DGV), Affymetrix Database of Genomic Variants (aDGV), Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER), Clinical Genome Resource (ClinGen), and Online Mendelian Inheritance in Man (OMIM). Cases with pathogenic and possibly pathogenic CNVs were classified as abnormal CMA, and those with normal CMA that included CNVs were classified as benign, possibly benign, or as a variation of unknown significance (VOUS).
The CNVs identified in the cases were tabulated using the Microsoft Excel program, and their classifications were verified in triplicate using visual parameters by analyzers trained at the FCM/Unicamp Cytogenetics and Cytogenomics Laboratory.
To describe the sample profile according to the variables under study, frequency tables of categorical variables with absolute frequency (n) and percentage (%) values were made, along with descriptive statistics of numerical variables, with mean values, standard deviation, minimum and maximum values, and median.
To compare categorical variables, Fisher’s exact test and the Mann–Whitney test were used for numerical variables. The significance level adopted was 5% (p < 0.05). For the statistical analysis, the following computer programs were used: The Statistical Analysis System (SAS) System for Windows v. 9.4 (SAS Institute Inc., 2002–2008, Cary, NC, United States).
ResultsFrom 1647 individuals with OC recorded in the BBCA until September 2018, 100 were selected for this study. The final sample comprised 44 participants clinically classified as NSOC and 56 as SOC. Their geographic origins were as follows: Northeast (65), Southeast (19), and South (16) of Brazil. Ages at selection varied from 0 to 51 years old.
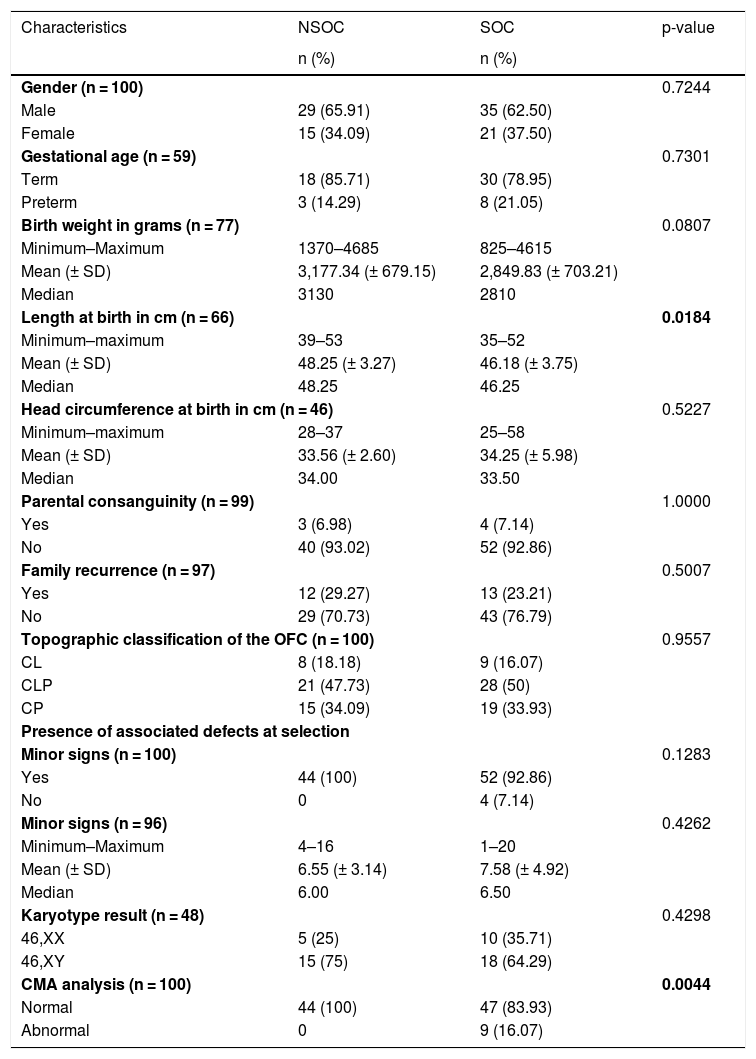
The comparison between the clinical characteristics of individuals with NSOC and SOC are presented in Table 1.
Comparison between the clinical characteristics of individuals with NSOC (n = 44 individuals) and SOC (n = 56 individuals).
| Characteristics | NSOC | SOC | p-value |
|---|---|---|---|
| n (%) | n (%) | ||
| Gender (n = 100) | 0.7244 | ||
| Male | 29 (65.91) | 35 (62.50) | |
| Female | 15 (34.09) | 21 (37.50) | |
| Gestational age (n = 59) | 0.7301 | ||
| Term | 18 (85.71) | 30 (78.95) | |
| Preterm | 3 (14.29) | 8 (21.05) | |
| Birth weight in grams (n = 77) | 0.0807 | ||
| Minimum–Maximum | 1370–4685 | 825–4615 | |
| Mean (± SD) | 3,177.34 (± 679.15) | 2,849.83 (± 703.21) | |
| Median | 3130 | 2810 | |
| Length at birth in cm (n = 66) | 0.0184 | ||
| Minimum–maximum | 39–53 | 35–52 | |
| Mean (± SD) | 48.25 (± 3.27) | 46.18 (± 3.75) | |
| Median | 48.25 | 46.25 | |
| Head circumference at birth in cm (n = 46) | 0.5227 | ||
| Minimum–maximum | 28–37 | 25–58 | |
| Mean (± SD) | 33.56 (± 2.60) | 34.25 (± 5.98) | |
| Median | 34.00 | 33.50 | |
| Parental consanguinity (n = 99) | 1.0000 | ||
| Yes | 3 (6.98) | 4 (7.14) | |
| No | 40 (93.02) | 52 (92.86) | |
| Family recurrence (n = 97) | 0.5007 | ||
| Yes | 12 (29.27) | 13 (23.21) | |
| No | 29 (70.73) | 43 (76.79) | |
| Topographic classification of the OFC (n = 100) | 0.9557 | ||
| CL | 8 (18.18) | 9 (16.07) | |
| CLP | 21 (47.73) | 28 (50) | |
| CP | 15 (34.09) | 19 (33.93) | |
| Presence of associated defects at selection | |||
| Minor signs (n = 100) | 0.1283 | ||
| Yes | 44 (100) | 52 (92.86) | |
| No | 0 | 4 (7.14) | |
| Minor signs (n = 96) | 0.4262 | ||
| Minimum–Maximum | 4–16 | 1–20 | |
| Mean (± SD) | 6.55 (± 3.14) | 7.58 (± 4.92) | |
| Median | 6.00 | 6.50 | |
| Karyotype result (n = 48) | 0.4298 | ||
| 46,XX | 5 (25) | 10 (35.71) | |
| 46,XY | 15 (75) | 18 (64.29) | |
| CMA analysis (n = 100) | 0.0044 | ||
| Normal | 44 (100) | 47 (83.93) | |
| Abnormal | 0 | 9 (16.07) | |
OC, typical oral cleft; CL, cleft lip; CLP, cleft lip and palate; CP, cleft palate; NSOC, non-syndromic oral cleft; SOC, syndromic oral cleft; CMA, chromosome microarray analysis.
There were no differences between NSOC and SOC with regard to clinical characteristics, except length at birth (p = 0.0184). Abnormal CMA was significant in SOC (p = 0.0044).
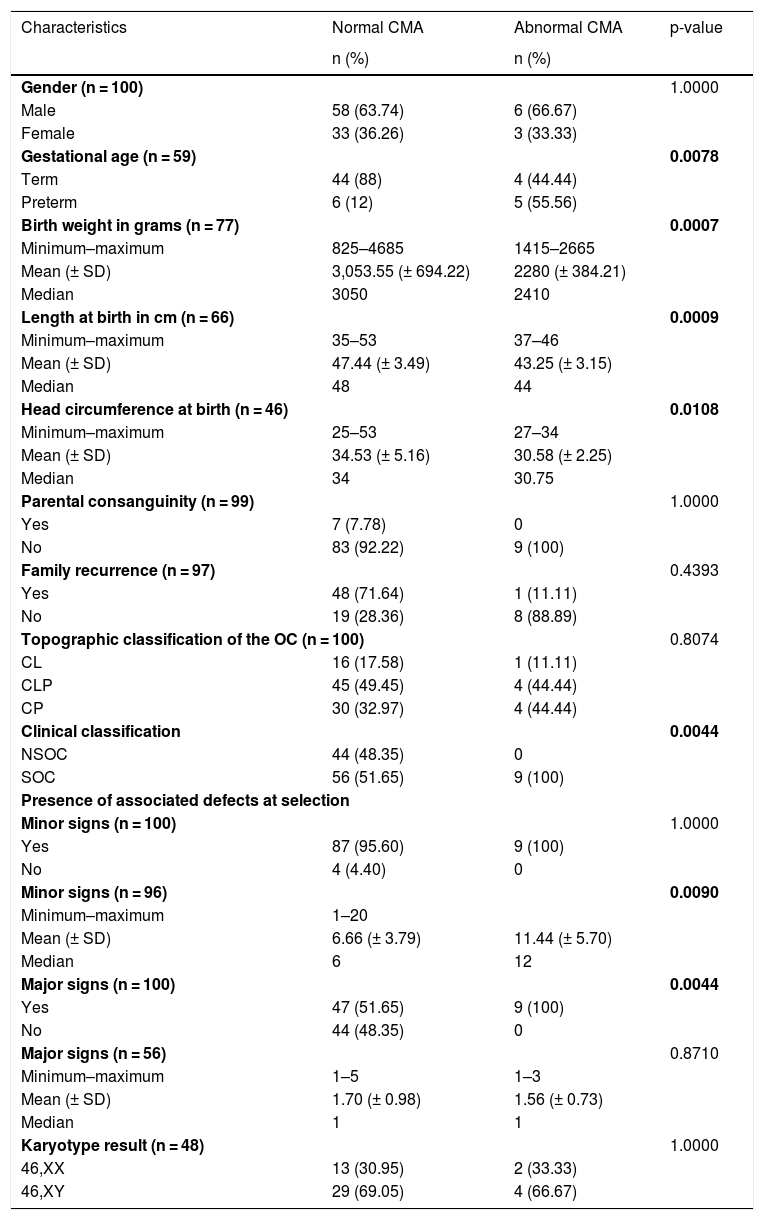
Table 2 illustrates the comparison between abnormal CMA and clinical characteristics.
Comparison between abnormal CMA and clinical characterization in the studied sample.
| Characteristics | Normal CMA | Abnormal CMA | p-value |
|---|---|---|---|
| n (%) | n (%) | ||
| Gender (n = 100) | 1.0000 | ||
| Male | 58 (63.74) | 6 (66.67) | |
| Female | 33 (36.26) | 3 (33.33) | |
| Gestational age (n = 59) | 0.0078 | ||
| Term | 44 (88) | 4 (44.44) | |
| Preterm | 6 (12) | 5 (55.56) | |
| Birth weight in grams (n = 77) | 0.0007 | ||
| Minimum–maximum | 825–4685 | 1415–2665 | |
| Mean (± SD) | 3,053.55 (± 694.22) | 2280 (± 384.21) | |
| Median | 3050 | 2410 | |
| Length at birth in cm (n = 66) | 0.0009 | ||
| Minimum–maximum | 35–53 | 37–46 | |
| Mean (± SD) | 47.44 (± 3.49) | 43.25 (± 3.15) | |
| Median | 48 | 44 | |
| Head circumference at birth (n = 46) | 0.0108 | ||
| Minimum–maximum | 25–53 | 27–34 | |
| Mean (± SD) | 34.53 (± 5.16) | 30.58 (± 2.25) | |
| Median | 34 | 30.75 | |
| Parental consanguinity (n = 99) | 1.0000 | ||
| Yes | 7 (7.78) | 0 | |
| No | 83 (92.22) | 9 (100) | |
| Family recurrence (n = 97) | 0.4393 | ||
| Yes | 48 (71.64) | 1 (11.11) | |
| No | 19 (28.36) | 8 (88.89) | |
| Topographic classification of the OC (n = 100) | 0.8074 | ||
| CL | 16 (17.58) | 1 (11.11) | |
| CLP | 45 (49.45) | 4 (44.44) | |
| CP | 30 (32.97) | 4 (44.44) | |
| Clinical classification | 0.0044 | ||
| NSOC | 44 (48.35) | 0 | |
| SOC | 56 (51.65) | 9 (100) | |
| Presence of associated defects at selection | |||
| Minor signs (n = 100) | 1.0000 | ||
| Yes | 87 (95.60) | 9 (100) | |
| No | 4 (4.40) | 0 | |
| Minor signs (n = 96) | 0.0090 | ||
| Minimum–maximum | 1–20 | ||
| Mean (± SD) | 6.66 (± 3.79) | 11.44 (± 5.70) | |
| Median | 6 | 12 | |
| Major signs (n = 100) | 0.0044 | ||
| Yes | 47 (51.65) | 9 (100) | |
| No | 44 (48.35) | 0 | |
| Major signs (n = 56) | 0.8710 | ||
| Minimum–maximum | 1–5 | 1–3 | |
| Mean (± SD) | 1.70 (± 0.98) | 1.56 (± 0.73) | |
| Median | 1 | 1 | |
| Karyotype result (n = 48) | 1.0000 | ||
| 46,XX | 13 (30.95) | 2 (33.33) | |
| 46,XY | 29 (69.05) | 4 (66.67) | |
OC, typical oral cleft; CL, cleft lip; CLP, cleft lip and palate; CP, cleft palate; NSOC, non-syndromic orofacial cleft; SOC, syndromic orofacial cleft; CMA, chromosome microarray analysis.
With regard to the CMA, there was a statistically significant difference in gestational age (p = 0.0078), mean weight (p = 0.0007), length (p = 0.0009), and head circumference at birth (p = 0.0108), which were lower in the group with abnormal CMA. Moreover, there was a significant predominance of abnormal CMA in individuals classified as SOC (p = 0.0044) as well. With regard to associated defects, the mean of minor signs (p = 0.0090) was statistically higher in cases with abnormal CMA, as well as the presence of major signs (p = 0.0044), which was one of the criteria for classification as SOC.
A total of 114 associated morphological defects were identified in the nine cases with abnormal CMA; additionally, there was variation in the topography of the distribution of associated defects: facial dysmorphisms 67/114 (58.77%), musculoskeletal 23/114 (20.17%), skin and appendages 9/114 (7.9%), genitourinary 8/114 (7.01%), cardiovascular 3/114 (2.63%), central nervous system (CNS) 2/114 (1.75%), and abdominal wall 2/114 (1.75%).
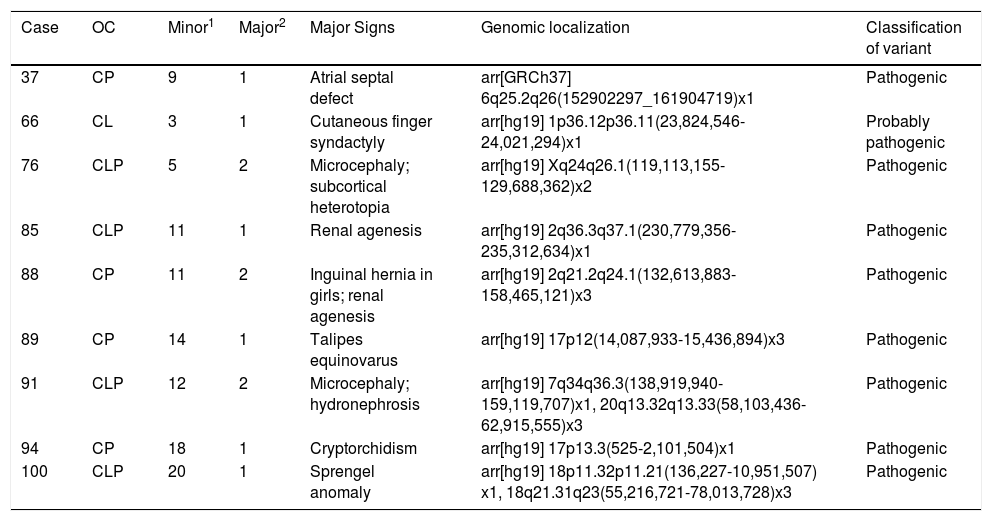
Table 3 depicts the topographic classification of the OC, number of associated signs, genomic locations, and the classification of the variants in cases with abnormal CMA.
Description of the topographic classification of the oral clefts, number of associated signs identified, genomic locations, and variant classifications in cases with abnormal CMA (n = 9).
| Case | OC | Minor1 | Major2 | Major Signs | Genomic localization | Classification of variant |
|---|---|---|---|---|---|---|
| 37 | CP | 9 | 1 | Atrial septal defect | arr[GRCh37] 6q25.2q26(152902297_161904719)x1 | Pathogenic |
| 66 | CL | 3 | 1 | Cutaneous finger syndactyly | arr[hg19] 1p36.12p36.11(23,824,546-24,021,294)x1 | Probably pathogenic |
| 76 | CLP | 5 | 2 | Microcephaly; subcortical heterotopia | arr[hg19] Xq24q26.1(119,113,155-129,688,362)x2 | Pathogenic |
| 85 | CLP | 11 | 1 | Renal agenesis | arr[hg19] 2q36.3q37.1(230,779,356-235,312,634)x1 | Pathogenic |
| 88 | CP | 11 | 2 | Inguinal hernia in girls; renal agenesis | arr[hg19] 2q21.2q24.1(132,613,883-158,465,121)x3 | Pathogenic |
| 89 | CP | 14 | 1 | Talipes equinovarus | arr[hg19] 17p12(14,087,933-15,436,894)x3 | Pathogenic |
| 91 | CLP | 12 | 2 | Microcephaly; hydronephrosis | arr[hg19] 7q34q36.3(138,919,940-159,119,707)x1, 20q13.32q13.33(58,103,436-62,915,555)x3 | Pathogenic |
| 94 | CP | 18 | 1 | Cryptorchidism | arr[hg19] 17p13.3(525-2,101,504)x1 | Pathogenic |
| 100 | CLP | 20 | 1 | Sprengel anomaly | arr[hg19] 18p11.32p11.21(136,227-10,951,507) x1, 18q21.31q23(55,216,721-78,013,728)x3 | Pathogenic |
OC, typical oral cleft; CL, cleft lip; CLP, cleft lip and palate; CP, palate cleft palate; 1 number of minor defects in selection; 2 number of major non-OC defects in selection.
Pathogenic CNVs were identified in four cases with CP and in four cases with CLP. The only case with CL presented the CNV classified as probably pathogenic. However, there was no statistical difference with regard to the topography of the OC (p = 0.8074).
DiscussionParticipants were selected from the BDCA, which employs a standard method to register clinical signs, thereby reducing the evaluator’s biases.22 A rigorous criterion was established to classify the individuals included in the SOC or NSOC groups. Therefore, although it appears to be a small sample, it is homogeneous and consistent.
It is expected that individuals with multiple defects as seen in SOC present restricted growth and prematurity due to embryological impairment and associated malformations.1 However, in the present study, the birth length was the only clinical characteristic that showed statistical difference in the comparison between NSOC and SOC. Although interesting in a pediatric scenario, this result should be interpreted with caution, as some differences in the technique of measurement of this variable may occur.
It should be noted that the same parameters at birth (prematurity, weight, length, and head circumference) had significantly lower results in the group of subjects with altered CMA. This result is in line with the expected intrauterine growth retardation in cases of multiple malformations. Thus, the evaluation of these parameters can help in the recognition of potentially syndromic cases and could be used as warning signs of genomic imbalances, including in cases with no major defects recognizable at physical examination.
Additionally, the mean of minor signs was statistically higher in cases with abnormal CMA, as well as the presence of major defects. The present study corroborates other reports, which state that the identification of minor signs has significant predictive value in diagnosing associated major defects and syndromic conditions.6,8
Morphological defects associated with OCs have been reported, with rates varying from 3% to 63%.5,25 The differences can be attributed to several factors, such as the examiner’s experience, patient’s age, OC severity, availability of diagnostic resources, inclusion/exclusion criteria, and clinical classification, among others. In 1987, Leppig et al. described that in 20% of the cases with three or more associated minor defects, a major sign was observed.8 In cases with abnormal CMA in this sample, the distribution of morphological defects is in accordance with the literature, although the frequency of diagnosis of CNS, cardiovascular disease, and genitourinary disorders was lower.25 The diagnosis of these abnormalities often requires complementary exams, which is essential for the proper phenotypic characterization of individuals with OC. However, in the face of the several healthcare needs that OC individuals require, these investigations are often postponed. For example, in Brazil, this is a limiting operational factor, as discussed by Gil-da-Silva-Lopes et al.14 This study emphasizes the importance of a detailed physical examination of the face and limbs, whose main signs are diagnosed by inspection.
There was also no difference in the karyotype results between NSOC and SOC. Chromosomal alterations are identified in approximately 50.7% of individuals with SOC and 0.9% in those with NSOC. The conventional karyotype can identify balanced chromosomal aberrations such as translocations and inversions, but usually recognizes abnormalities greater than 10 Mb, and its sensitivity is lower than the CMA (3.6% vs. 7.7%).20,26 In this sample, 48 cases were analyzed with karyotype, and all had normal results. Of the nine cases with abnormal CMA, three cases (37, 66, and 85) had not been previously studied with karyotype; however, the alterations presented were smaller than 5 Mb and possibly would not be detected by conventional cytogenetics.
The use of the CMA technique demonstrates the ability to detect pathogenic CNVs in approximately 14–18% of cases with developmental delay, intellectual disabilities, and multiple congenital malformations.4,19 In the present study, abnormal CMA was observed in 9% of the cases, all the cases that were previously classified as SOC. Although few studies describe the CMA technique for OC investigation, they are not comparable as different designs were used. Within those cases, the abnormal CMA varies from 1.92% to 78.57% in cases classified as NSOC and 1.58% to 71.42% in SOC.4,16–21
In individuals with abnormal CMA (Table 3), some interesting aspects can be highlighted.
In individual 37, male, the presented phenotype comprises an incomplete cleft palate and a complete soft palate cleft (HP: 0410005, HP: 0000185), atrial septal defect (HP: 0001631), and nine minor signs. The CMA analysis identified a pathogenic deletion of 8.95 Mb located at 6q25.2q26; this region comprises a 6q24-q25 microdeletion syndrome, contiguous gene deletion syndrome (OMIM # 612863), and is compatible with the descriptions of 6q25.2q26 microdeletion as reported in the literature.
Individual 66, female, presented a left cleft lip (HP: 0410030), cutaneous finger syndactyly (HP: 0010554), and three minor signs. The CMA analysis identified a probably pathogenic 197 Kb deletion located at 1p36.12p36.11. The 1p36 microdeletion syndrome is a contiguous gene deletion syndrome (OMIM # 607872) with several changes not found in the present case. However, the variant found contains only four OMIM genes, including the Diamond-Blackfan anemia type 7 ribosomal protein l11 [RPL11] gene (# 612562), which is characterized by intrauterine growth retardation, delayed growth, facial dysmorphisms, cleft palate, heart disease, and thumb deformity. Quite possibly, because the deleted variant in case 66 contains only four OMIM genes, the phenotype presented is not characteristic of 1p36 microdeletion syndrome, except for the OC. It is possible that cleft lip and hand abnormalities may be justified by the deletion involving the Diamond-Blackfan anemia type 7-related ribosomal protein l11 [RPL11]. Complementary examinations of the mother who has thumb duplication are still required.
Individual 88, female, showed complete cleft palate (HP: 0000175), inguinal hernia (HP: 0000023) – which is considered as major sign in girls, renal agenesis (HP: 0000104), and eleven minor signs. The CMA analysis revealed a pathogenic duplication of approximately 25.85 Mb in 2q21.2q24.1 containing 48 OMIM genes. Although there are no reported changes with the exact size to those presented in this case in the ClinVAr and DECIPHER bases, the phenotype is compatible with the descriptions of larger or smaller size variants being referred: developmental delay and minors signs (nssv582319; nssv582710; nssv706887, 254507; 274392; 282249; 305834; 363398; 363398); hearing impairment (248386); renal abnormalities, atrial septal defect, late cranial suture closure, and short stature (251953). However, in view of the gestational history, the involvement of additional teratogenic agents in this phenotype cannot be ruled out, as there is a report of alcohol consumption and Cannabis sativa use during pregnancy.
Individual 89, female, manifests incomplete cleft soft palate (HP: 0000185) and talipes equinovarus (HP: 0001762), in addition to 14 minor defects. The CMA technique revealed a pathogenic duplication of approximately 1.34 Mb in 17p12 containing four OMIM genes. It is possible that the neuropsychomotor developmental delay, behavioral changes, and foot deformities may be justified by the duplication involving the peripheral myelin protein 22 gene [PMP22] (*601097), this duplication is classically found in patients with Charcot-Marie-Tooth.
Individual 91, male, has a bilateral cleft lip and palate (HP: 0002744), microcephaly (HP: 0000252), hydronephrosis (HP: 0000126), and 12 minor signs. The phenotype presented is corroborated by the descriptions in the literature with regard to the two pathogenic variants identified in the analysis of CMA (7q34q36.3 deletion and 20q13.32q13.33 duplication).
Cases 76, 85, 94, and 100 were previously described.14,27,28
This study established the importance of applying the CMA technique to identify chromosomal imbalances in SOC individuals, in addition to dysmorphological evaluation. These are relevant and universal issues in clinical practice, especially for intervention, care, and genetic counseling. Etiological investigation and genetic counseling are important especially when associated anomalies are presented.
The CMA technique contributed to the etiological elucidation of the SOC cases, which is essential for proper management and genetic counseling. The presence of OC with prematurity, low weight, length, and head circumference at birth, as well as the identification of minor defects, should draw the evaluator’s attention for etiological investigation. Follow-up would be useful for identification of unapparent major features and/or neurodevelopmental delay. This approach favors the diagnosis of syndromic conditions at an appropriate time, therapeutic planning, global rehabilitation, and the child’s social insertion.
FundingThis study was supported by the São Paulo Research Foundation (FAPESP#2012/51799-6) and the Alagoas Research Foundation (FAPEAL#60030 000707/2013). VLGSL receives support from CNPq#305985/2017-5; TMF receives support from CNPq #306245/2016-7.
Ethics approval and consent to participateThis study was approved by the Unicamp Research Ethics Committee (CAAE 35316314.9.1001.5404).
Informed consentInformed consent was obtained from all individual participants included in the study or from their parents.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank the participant individuals and their relatives and the Department of Statistics, Faculty of Medical Science, State University of Campinas (Unicamp), Campinas, SP, Brazil. This study was supported by the São Paulo Research Foundation (FAPESP #2012 / 51799-6) and the Alagoas Research Foundation (FAPEAL #60030 000707/2013). VLGSL receives support from CNPq #305985 / 2017-5; TMF receives support from CNPq #306245/2016-7.




