


Arthrogryposis multiplex congenita is a relatively rare neuromuscular syndrome, with a prevalence of 1:3000–5000 newborns. In this study, the authors describe the clinical features of a group of 50 unrelated Mexican patients with arthrogryposis multiplex congenita.
MethodsPatients were diagnosed by physical and radiographic examination and the family history was evaluated.
ResultsOf the 50 cases, nine presented other features (pectum excavatum, cleft palate, mental retardation, ulnar agenesis, etc.). Environmental factors, as well as prenatal and family history, were analyzed. The chromosomal anomalies and clinical entities associated with arthrogryposis multiplex congenita were reported. No chromosomal aberrations were present in the cases with mental retardation. Three unrelated familial cases with arthrogryposis multiplex congenita were observed in which autosomal recessive, autosomal dominant and X-linked inheritance patterns are possible. A literature review regarding arthrogryposis multiplex congenita was also conducted.
ConclusionsIt is important to establish patient-specific physical therapy and rehabilitation programs. A multidisciplinary approach is necessary, with medical, surgical, rehabilitation, social and psychological care, including genetic counseling.
A Artrogripose múltipla congênita é uma síndrome neuromuscular relativamente rara, com prevalência de 1:3000-5000 recém-nascidos. É por isso que, neste estudo, descrevemos as características clínicas de um grupo de 50 casos de pacientes mexicanos não relacionados com Artrogripose múltipla congênita.
MétodosOs pacientes foram diagnosticados por exame físico e radiográfico, e o histórico familiar foi avaliado.
ResultadosDescrevemos 50 pacientes não relacionados com Artrogripose múltipla congênita.
Nove deles apresentaram outras características (pectus excavatum, fissura palatina, retardo mental, agenesia da ulna, etc.). Foram analisados os fatores ambientais, pré-natais e o histórico familiar. Relatamos as anomalias cromossômicas e as entidades clínicas associadas com a Artrogripose múltipla congênita. Não havia nenhuma aberração cromossômica nos casos com retardo mental. Também encontramos 3 casos familiares não relacionados com Artrogripose múltipla congênita, em que são possíveis padrões de herança autossômica recessiva, autossômica dominante e ligada ao cromossomo X. Também analisamos a preocupação da literatura com a Artrogripose múltipla congênita.
ConclusõesReiteramos a ideia de que é importante estabelecer programas de fisioterapia e reabilitação específicos para os pacientes. É necessária uma abordagem multidisciplinar com cuidado médico, cirúrgico, de reabilitação, social e psicológico, incluindo aconselhamento genético.
The term arthrogryposis is derived from the Greek words “arthron” and “gryposis”, which mean “curved joints”. Multiple synonyms have been used to describe this clinical feature: multiple congenital articular rigidity, amyoplasia congenital, myodystrophia fetalis deformans, congenital arthromyodysplasia, multiple congenital contractures and arthrogryposis multiplex congenita (AMC).1,2 AMC refers to a large heterogeneous group of conditions, characterized by contracted muscles and non-progressive congenital limitation of movement of two or more different joints with thick periarticular capsules. AMC is relatively rare, occurring in approximately 1:3000–5000 newborns; males and females are equally affected. All four extremities are involved in 50–60% of cases; lower limbs, in 30–40%, and upper limbs, in 10–15%. In 20% of cases, there is dislocation and subluxation of hips, patellas and knees, while vertebral and temporomandibular joints are rarely involved.3,4 The most common type of AMC is amyoplasia, which represents one-third of all cases. Amyoplasia is characterized by symmetrical positioning of the limbs with severe talipes equinovarus and extended elbows. A feature that often occur Sis a characteristic midline facial hemangioma. Some patients could also have abdominal structure or genital abnormalities, downsloping and internally rotated shoulders, and pronated forearms with flexion deformities of wrists and fingers. In addition to talipes equinovarus, lower limb involvement most often includes flexed or extended knees; hips may present as flexed and externally rotated, extended and subluxated, or dislocated.3,5,6
AMC can be present in over 150 specific conditions, which include a large group of myopathic and neurogenic disorders, connective tissue anomalies, and factors that produce limitation of fetal motion (structural abnormalities of the uterus, amniotic bands syndrome, twining, and oligo or polyhydramnios).7,8 In animal models, contractures have been induced by viruses, neuromuscular blocking, insecticides, anticonvulsants, neuromuscular blocking agents, ethanol, limb immobilization, and hyperthermia.8–14 These factors appear to interfere with the development of the limbs, resulting in loss of muscle mass with imbalance of muscle power at the joints. The etiology of AMC includes both genetic and environmental factors; among the genetic causes, single gene defects and chromosomal disorders have been reported. Molecular analysis has revealed that a determined phenotype can be caused by mutations in different genes, evidencing the genetic heterogeneity of this disease.3
The present study aimed to describe the clinical features of 50 cases with AMC, including a brief review of the literature regarding this topic.
Patient and methodsA prospective and cross-sectional study was conducted with 50 patients with AMC. These patients were referred to the Genetics Department of the National Institute of Rehabilitation (Mexico City, Mexico) with presumptive diagnosis of AMC, during a period of three years. All were Mexican descendants. Patients and their parents were informed about the characteristics of the study and agreed to participate.
The study's protocol was approved by the Institute's research and ethics committees. The diagnosis of AMC was based on typical clinical features, such as non-progressive joint contractures limiting extension or flexion movements evident at birth, involving more than one joint, either from the upper or lower limbs or both. In all cases, the patients were subjected to physical and radiographic examination. The medical family history and delivery data were assessed during medical consultation in the department of Genetics. Pregnancy history evaluation included position of the fetus in utero, infections, fever, use of illegal substances, medications, and unusual events during pregnancy. Karyotype analysis was performed from peripheral blood leukocytes using G banding stain according to standard techniques. Briefly, peripheral blood collected by venous puncture, using a heparinized syringe, was cultivated in the presence of phytohaemagglutinin. After 72h, cell growth was stopped by adding colcemid. Cell cultures were harvested and treated with hypotonic and fixative solution. Samples were placed on glass slides for staining15; 25 metaphases were analyzed under a light microscope for each case.
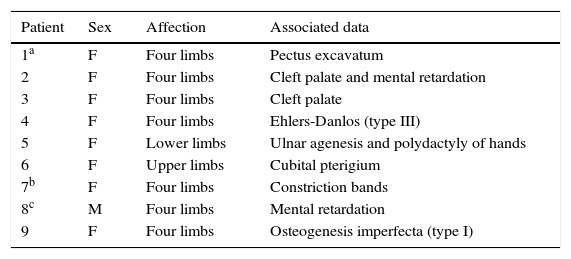
ResultsIn the present study, 28 (56%) patients were females and 22 (44%) males; both sexes were affected. 27 (54%) patients were affected in all four limbs; lower and upper limbs were affected in 18 (36%) and five (10%) cases respectively. The distribution of limb involvement is shown in Fig. 1A. Articular compromise can present a great variability, even within the same patient: one joint can be completely extended while the contralateral joint is in flexion. Fig. 1B depicts a clinical involvement of upper limbs: forearm pronosupination was compromised bilaterally, wrists and interphalangeal joints were fixed in a semi-flexed position and no flexion creases were present. Fig. 1C exemplifies lower limbs involvement, where the right and left knees were fixed in the semi-flexed and extended position, respectively; the right ankle was extended and the left ankle was in adduction, and the patient was unable to move them. The medium age at presentation to the clinic was 4.6 years (age range from 0 to 7 years). When investigating the involvement of other joints, hip dislocation and subluxation was observed in 14 cases (28%), and joint hypermobility with hyperflexion were present in two cases. A great variety of congenital abnormalities (orthopedic or non-orthopedic) are reported to be associated with AMC. In this cohort, other clinical features sporadic presentation was also observed in nine out of 50 cases (Table 1). In the present series, maternal disease was present in ten cases: one case with diabetes mellitus, five cases presented urinary tract infection and fever (above 39°C) in the first and second trimester of pregnancy, four cases of eclampsia, and six had vaginal bleeding during the first trimester. Delivery history indicated that fetal position at delivery was cephalic in 25 cases, breeched in 16, and transverse in seven cases; 2 cases had other positions. One multiple birth (dizygotic twins) and a case with uterus abnormalities were also observed.
 studied. (A) Percentage of involvement of limbs. (B) Upper limbs from an affected patient; forearm pronosupination movements were not possible, wrists and interphalangeal joints were fixed in semi-flexion position and no flexion creases were observed. (C) Lower limbs of a patient with AMC; right and left knees were fixed in semi-flexion and extension position respectively, right ankle was extended and the left ankle in adduction.")
General characteristics of the patients with arthrogryposis multiplex congenita (AMC) studied. (A) Percentage of involvement of limbs. (B) Upper limbs from an affected patient; forearm pronosupination movements were not possible, wrists and interphalangeal joints were fixed in semi-flexion position and no flexion creases were observed. (C) Lower limbs of a patient with AMC; right and left knees were fixed in semi-flexion and extension position respectively, right ankle was extended and the left ankle in adduction.
Congenital anomalies associated with arthrogryposis multiple congenita.
| Patient | Sex | Affection | Associated data |
|---|---|---|---|
| 1a | F | Four limbs | Pectus excavatum |
| 2 | F | Four limbs | Cleft palate and mental retardation |
| 3 | F | Four limbs | Cleft palate |
| 4 | F | Four limbs | Ehlers-Danlos (type III) |
| 5 | F | Lower limbs | Ulnar agenesis and polydactyly of hands |
| 6 | F | Upper limbs | Cubital pterigium |
| 7b | F | Four limbs | Constriction bands |
| 8c | M | Four limbs | Mental retardation |
| 9 | F | Four limbs | Osteogenesis imperfecta (type I) |
F, female; M, male.
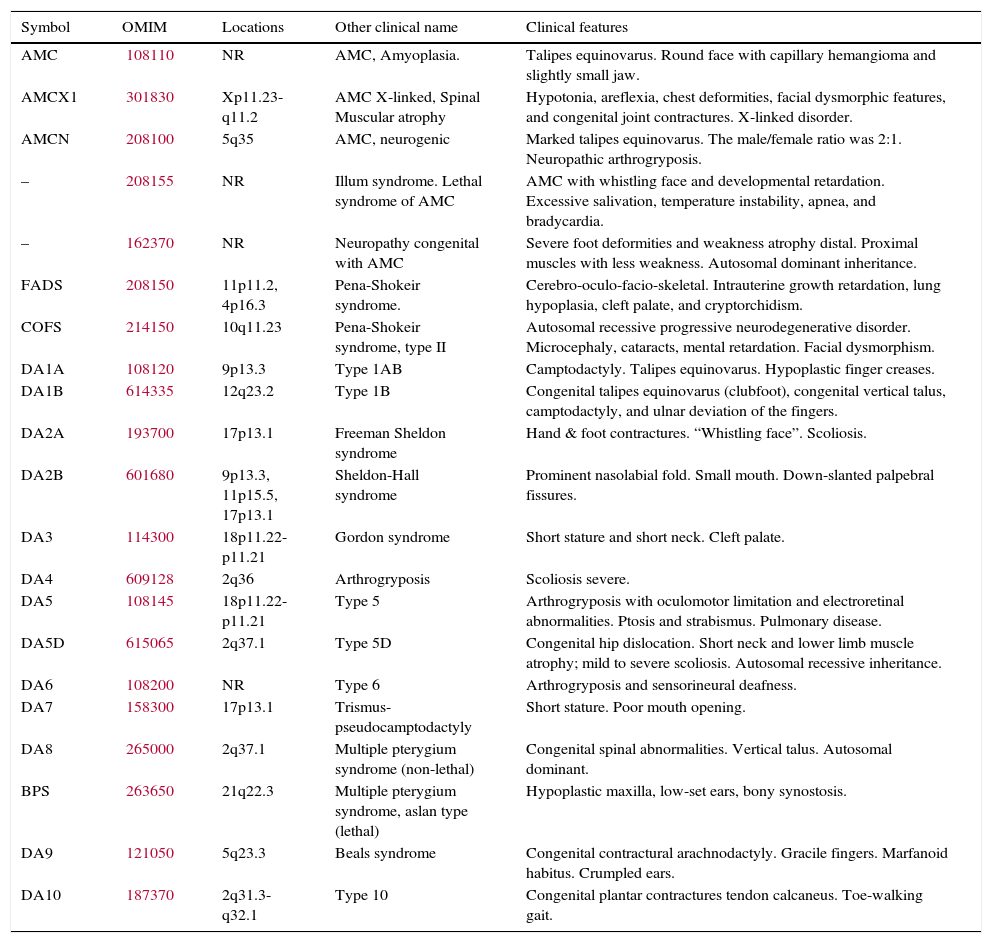
Table 2 presents a brief review of the literature about different variants of AMC and distal arthrogryposis. In the present series, there were two unrelated non-familial cases where consanguinity was observed. In addition, three familial cases with no evidence of consanguinity were also observed (Fig. 2).
Some different variants described in the classification of multiplex congenital and distal arthrogryposis.
| Symbol | OMIM | Locations | Other clinical name | Clinical features |
|---|---|---|---|---|
| AMC | 108110 | NR | AMC, Amyoplasia. | Talipes equinovarus. Round face with capillary hemangioma and slightly small jaw. |
| AMCX1 | 301830 | Xp11.23-q11.2 | AMC X-linked, Spinal Muscular atrophy | Hypotonia, areflexia, chest deformities, facial dysmorphic features, and congenital joint contractures. X-linked disorder. |
| AMCN | 208100 | 5q35 | AMC, neurogenic | Marked talipes equinovarus. The male/female ratio was 2:1. Neuropathic arthrogryposis. |
| – | 208155 | NR | Illum syndrome. Lethal syndrome of AMC | AMC with whistling face and developmental retardation. Excessive salivation, temperature instability, apnea, and bradycardia. |
| – | 162370 | NR | Neuropathy congenital with AMC | Severe foot deformities and weakness atrophy distal. Proximal muscles with less weakness. Autosomal dominant inheritance. |
| FADS | 208150 | 11p11.2, 4p16.3 | Pena-Shokeir syndrome. | Cerebro-oculo-facio-skeletal. Intrauterine growth retardation, lung hypoplasia, cleft palate, and cryptorchidism. |
| COFS | 214150 | 10q11.23 | Pena-Shokeir syndrome, type II | Autosomal recessive progressive neurodegenerative disorder. Microcephaly, cataracts, mental retardation. Facial dysmorphism. |
| DA1A | 108120 | 9p13.3 | Type 1AB | Camptodactyly. Talipes equinovarus. Hypoplastic finger creases. |
| DA1B | 614335 | 12q23.2 | Type 1B | Congenital talipes equinovarus (clubfoot), congenital vertical talus, camptodactyly, and ulnar deviation of the fingers. |
| DA2A | 193700 | 17p13.1 | Freeman Sheldon syndrome | Hand & foot contractures. “Whistling face”. Scoliosis. |
| DA2B | 601680 | 9p13.3, 11p15.5, 17p13.1 | Sheldon-Hall syndrome | Prominent nasolabial fold. Small mouth. Down-slanted palpebral fissures. |
| DA3 | 114300 | 18p11.22-p11.21 | Gordon syndrome | Short stature and short neck. Cleft palate. |
| DA4 | 609128 | 2q36 | Arthrogryposis | Scoliosis severe. |
| DA5 | 108145 | 18p11.22-p11.21 | Type 5 | Arthrogryposis with oculomotor limitation and electroretinal abnormalities. Ptosis and strabismus. Pulmonary disease. |
| DA5D | 615065 | 2q37.1 | Type 5D | Congenital hip dislocation. Short neck and lower limb muscle atrophy; mild to severe scoliosis. Autosomal recessive inheritance. |
| DA6 | 108200 | NR | Type 6 | Arthrogryposis and sensorineural deafness. |
| DA7 | 158300 | 17p13.1 | Trismus-pseudocamptodactyly | Short stature. Poor mouth opening. |
| DA8 | 265000 | 2q37.1 | Multiple pterygium syndrome (non-lethal) | Congenital spinal abnormalities. Vertical talus. Autosomal dominant. |
| BPS | 263650 | 21q22.3 | Multiple pterygium syndrome, aslan type (lethal) | Hypoplastic maxilla, low-set ears, bony synostosis. |
| DA9 | 121050 | 5q23.3 | Beals syndrome | Congenital contractural arachnodactyly. Gracile fingers. Marfanoid habitus. Crumpled ears. |
| DA10 | 187370 | 2q31.3-q32.1 | Type 10 | Congenital plantar contractures tendon calcaneus. Toe-walking gait. |
AMC, arthrogryposis multiplex congenital; DA, distal arthrogryposis; FADS, fetal akinesia deformation sequence; COFS, cerebrooculo-facio-skeletal syndrome; BPS, Bartsocas-Papas syndrome; OMIM, Online Mendelian Inheritance in Man; NR, not reported.
 with the same type of Arthrogryposis multiplex congenital. Arrows indicate the affected patients. Squares represent males and circles, females; filled squares/circles represent affected individuals.")
In AMC, a contracture is the limitation of mobility at a specific joint; it does not represent an intrinsic problem of the joint during fetal development, because joint organization appears normal. It is the lack of joint mobility during the fetal growth, which is associated with the development of extra connective tissue around the joints. These deformities are usually symmetric, non-progressive, and involve more than one body area.3 AMC is very evident at birth, and diagnosis is possible prenatally with real-time ultrasound.16,17
In AMC, all four extremities are involved in 50–60% of cases; lower limbs, in 30–40%, and upper limbs, in 10–15%.7 In this series, 54% of the patients showed involvement of all extremities, 36% of the lower limbs, while the upper limbs were involved in 10%; no differences between males and females were observed. These data agree with those reported in the international literature (Fig. 1).3,7,13,14,18
At least 150 specific clinical entities have been described with AMC. It is apparent that multiple etiologic factors converge upon one common response, which results in loss of movement of the joints. AMC is also known to occur as a component of a large group of heterogeneous myopathic, neurogenic, and vascular disorders, as well as other anomalies such as skeletal dysplasia.3,7Table 2 depicts an updated summary of different variants described in the classification of AMC and distal arthrogryposis. Abnormal connective tissue, specifically collagen, may interfere with the normal development of bone, cartilage, and tendons; this can result in joint contractures and fibrosis. In this series, one patient had Ehlers–Danlos type 3 and one had osteogenesis imperfecta type I (patients 4 and 9, Table 1).
Prenatal history is very important in the study of AMC. In this cohort, maternal diseases such as diabetes mellitus, urinary tract infection with fever and eclampsia were observed. There was also one event of vaginal bleeding during the first trimester of pregnancy. The identification of these environmental and sporadic non-genetic factors is important to establish specific types of AMC. It is possible that environmental factors have contributed in the etiology of AMC in these cases.
It has been described that abnormal fetal positions, multiple gestation, and structural abnormalities of the uterus can produce limitation of the joint movements and consequently promote AMC. Half of the present patients had cephalic presentation while the other half showed one of the considered risk presentations (breeched, transverse, or other position). One multiple birth and one case with uterus abnormalities, which are also considered as risk factors, were also observed.
Specific inheritance modes of AMC are described for particular forms of contractures. Sometimes AMC is caused by a defect in a single gene; in these cases, autosomal dominant, autosomal recessive and X-linked inheritance is possible. At the moment, several loci related with different types of AMC have been reported, for example: contractural arachnodactyly (5q23-31), diastrophic dysplasia (5q31), lethal spinal muscular atrophy (5q13.3), distal arthrogryposis type I (9q21.2), symphalangism (9q), lethal X-linked arthrogryposis (Xp11.3-q11.2), and ophthalmoplegia (12p11.2-q12) (Table 2).10–13,19–24 There are also some reports where a patient whit AMC carries a duplication in a chromosome and a deletion in another chromosome.23
Familial analysis is an important part of the evaluation of a child with AMC – it is necessary in order to suggest the inheritance mode and provide recurrence risk to the parents. Fig. 2A presents a family tree with three affected siblings (hands and feet). Although there was no data of consanguinity, the most probable trait of transmission is autosomal recessive; germline mosaicism is another possibility that cannot be ruled out. Fig. 2B presents a family tree with two affected siblings (hands and feet). Autosomal recessive or X-linked traits are the most probable mode of transmission, although germline mosaicism could also be considered. Fig. 2C presents a family tree with three affected members in three generations (hands and feet). This genealogy strongly suggests an autosomal dominant inheritance. The fact that patient II-1 is not affected can be explained due to incomplete penetrance.
AMC may also be caused by numerical or structural chromosomal aberrations. These abnormalities are particularly frequent in cases of AMC associated with mental retardation. Most of these chromosome abnormalities are present as de novo events, which means that the risk of recurrence for relatives is very low.25,26 In this study, chromosomal abnormalities were present in two out of 50 cases, one with 47,XY,+21 and another one with 47,XXY. Nevertheless, these chromosomal anomalies are not associated with AMC. The two patients with mental retardation in this series (2 and 8) did not present chromosomal anomalies, as showed by karyotype analysis performed in peripheral blood leukocytes. However, the possibility of a chromosomal mosaicism in a small proportion cannot be discarded, as no other tissue, such as skin (fibroblast), was used for karyotype analysis.25 Chromosomal studies in patients with AMC and mental retardation or multisystemic disorders are very important for the recognition of clinical syndromes with chromosomal implication.
The postnatal diagnosis of AMC requires a compilation of medical records, family history (i.e., other family members affected, hyperextensibility, hypotonia, and other signs related to the disease), laboratory data (serum enzymes), radiographic analysis, electrophysiological studies, and pathologic studies (including muscle and nerve biopsy). For cases considered to be sporadic, where no relevant medical history is found and no mendelian inheritance pattern can be observed, the empiric risk of recurrence is around 3–5%. The appropriate treatment in AMC should be multidisciplinary, with the participation of a pediatrician with specialty in orthopedics, a physical therapist, a geneticist, and a neurologist, among others. The primary objective of the treatment is to improve the function of the affected limbs. The management of potentially preventable complications (joint dislocation, osteoarthritis, and scoliosis) is fundamental to ensure the quality of life of the patients. Conversely, rehabilitation programs facilitate and promote independent function in everyday activities. And finally, as has been exposed above, this condition involves genetic and environmental factors; therefore, genetic counseling should be taken into consideration if the family plans to have another child.27,28
In this article, the classification of AMC was updated according to the literature. Only a few reports on AMC in Mexican population were retrieved. In 1976, a report described 83 cases29; 46 cases were reported in 1991,30 and a manuscript related to distal arthrogryposis type IIB was published in 1999.22 In this study, 50 new cases were included, with variable degree of severity. Some of these cases were associated with other medical problems, which resulted in severe limitations in these patients. Therefore, it is important to establish patient-specific physical therapy and rehabilitation programs, according to the particular characteristics and evolution of each case. As in previous studies, the authors reiterate that it is necessary a multidisciplinary approach, with medical, surgical, rehabilitation, social, and psychological care, including genetic counseling, in order to achieve a successful management of these patients.
Conflicts of interestThe authors declare no conflicts of interest.
These authors contributed to the preparation of this manuscript in the same way.
Please cite this article as: Valdés-Flores M, Casas-Avila L, Hernández-Zamora E, Kofman S, Hidalgo-Bravo A. Characterization of a group unrelated patients with arthrogryposis multiplex congenita. J Pediatr (Rio J). 2016;92:58–64.




