
This minireview gathers the scientific foundations of the literature on genetic errors in the development of the humoral immune system to help pediatricians suspect these defects.
SourcesA systemic search using the PubMed MEDLINE database was performed for all Predominantly Antibody Deficiencies (PADs) described in the 2020 IUIS Expert Committee for PID classification system, combined with terms for hypogammaglobulinemia. Search terms for PADs were based on the listed names and affected genes as classified by the IUIS 2020. Abstracts of the results were reviewed to find relevant case series, review articles of PADs associated with infection, opportunistic infection, autoimmunity, cytopenias, malignancies, inflammatory diseases, neurological and respiratory diseases. References from relevant articles were further reviewed for additional references. Relevant findings were grouped in accordance with the IUIS 2020 classification system. Clinical and genetic features, if known, were described.
Data synthesisPADs refer to impaired antibody production due to molecular defects intrinsic to B cells or a failure of interaction between B and T cells. The patients develop recurrent or chronic infection or respond to the antigens with dysregulation of the immune function, causing severe allergy, autoimmunity, inflammation, lymphoproliferation and malignancy. The diagnosis is a combined exercise of clinical and laboratory investigation similar to that performed by Bruton (1952). In the context of SARS-CoV-2 infection, the experience of XLA and CVID patients has been surprising. Variants in 39 genes were reported as causing PADs, but the clinical heterogeneity within each variant is not clear.
ConclusionBruton (1952) used clinical expertise and protein electrophoresis to identify XLA. The IUIS (2020) committee used immunoglobulins and B lymphocyte to characterize PADs. Pediatricians should suspect it to detect it and prevent morbidities that can have an astonishing and irreversible impact on the child's life.
Over 430 distinct genetic abnormalities result in Human Inborn Errors of Immunity (HIEI)1 and represent “An expanding universe”.2 Hypogammaglobulinemia is a feature of several forms of HIEI, including Predominantly Antibody Deficiencies (PADs) and Combined Immunodeficiency affecting infants and young children. A global systematic review of PID registries (104,614 patients)3 and the Jeffrey Modell Centers Network (JMCN) global report (187,988 patients)4 agree that PADs (51.9% vs. 45.1%, respectively) and combined immunodeficiency (11.8% vs. 6.1%, respectively) are the most often reported HIEI in the world. The number of males affected is higher than females (5 vs.1.3), highlighting the X-linked disorders. When consanguinity is present, the frequency of AR increases.3,4 With rare exceptions, PADs are not obvious at birth but become evident when the affected individual is exposed to pathogenic microorganisms and develops recurrent or chronic infection, or responds to the antigens with dysregulation of immune function causing severe allergy, autoimmunity, inflammation, lymphoproliferation, and malignancy.5–8 As the immune system has high connectivity and is present in all tissues, it is natural that the infectious and non-infectious manifestations of the genetic errors of the immune system can be revealed in any tissue (hematopoietic, gastrointestinal, respiratory, osteoarticular, muscular, skin, central nervous system and peripheral nervous system) and at any age. The pediatrician has the chance to be the first to suspect primary immunodeficiency through family history, infectious history, the number of neutrophils and lymphocytes in the blood count and the thymic image when there is a need to perform chest X-rays. The opportunities for this diagnostic suspicion continue in each clinical childcare consultation and in each lab test result performed.
The B-cell lineage defect is inherent to the B cell itself, but is often secondary to abnormal T lymphocytes, which cannot provide adequate signals for B lymphocytes to thrive. Thus, as a rule, altered B-cell population accompanies T-lymphocyte abnormalities.9–12
The patients may also have serious adverse events with live strain vaccines (such as yellow fever, polio, measles, mumps, rubella and rotavirus) or by acquiring infections from healthy individuals who have not been immunized or who are shedding live vaccine-derived viruses.5–8,13 During the yellow fever vaccination campaign (2018), a 10-month-old boy developed a yellow fever vaccine-associated neurological disease as the first sign of XLA (personal communication). Early diagnosis with newborn screening makes it possible to prevent these complications. Screening programs utilize TREC assay for SCID, and B cell lymphopenia can be detected through quantification of KREC.14 Furthermore, the form of gene therapy is a hope for potential cure for these patients.7
Most frequencies of PADs3,4,8 show a varied prevalence worldwide, and its clinical profile will also vary,12,15 as well as the phenotype in patients with similar variants.16 The International Union of Immunological Societies’ Inborn Errors of Immunity Committee (IUIS-IEI) reported 39 genes causing PADs and assembled the disorders according only to the serum immunoglobulin levels and B-cell number:1
- 1.
Severe reduction in all serum immunoglobulin isotypes with profoundly decreased or absent B cells, agammaglobulinemia;
- 2.
Severe reduction in at least 2 serum immunoglobulin isotypes with normal or low number of B cells, CVID phenotype;
- 3.
Severe reduction in serum IgG and IgA with normal/elevated IgM and normal numbers of B cells, hyper IgM;
- 4.
Isotype, light chain, or functional deficiencies with generally normal numbers of B cells.
PADs consist of a phenotype's spectrum that ranges from the relatively mild, such as IgAD and THI, to those with severe antibody loss, as in XLA, CVID, and HIGM.5,10,17 The most prevalent symptomatic form of PAD is CVID, defined by profoundly low IgG and IgA and/or IgM as well as failure to mount antibodies against vaccination.12,17,18 The term “variable common immunodeficiency” was created by WHO and redefined in 2009 by the Primary Immunodeficiency Diseases (PID) Classification Committee of the IUIS.17 Patients with PADs have recurrent infections with encapsulated bacteria or a history of failure to respond to antibiotic treatment. However, those individuals affected by SIgAD or THI may have few or no infections.10 This minireview, which is limited in scope, provides an overview of the clinical features of the XLA, CVID, HIGM, SigAD, and THI.
Severe reduction in all serum immunoglobulin isotypes with profoundly decreased or absent B cells, agammaglobulinemiaIn this group, XLA9,10,19,20 and ARA10,18 are caused by a differentiation defect inherent to the B-cell lineage in bone marrow, resulting in very low or absence of serum IgM, IgG, IgA, IgE levels, absent or very low B-lymphocyte <1–2% (CD19) and failure of antibody production to vaccines.9,10,19,20 The tonsils, spleen, adenoids, Peyer's patches and lymph nodes where B cells undergo maturation, differentiation, and storage have reduced size.[7,10]
Phenotypically, ARA and XLA are often indistinguishable but different inheritance. Both are highly suscetible to infections with encapsulated bacteria but also to enteroviruses.10,21,22
X-linked agammaglobulinemia (XLA)XLA is caused by defects in Btk (gene) encoding Bruton Tyrosine Kinase (Btk) and accounts for 85% of cases of congenital agammaglobulinemia.10,19,20 The estimated incidence ranges from 1:100,000 to 1:200,000 live births.8–10 BTK is expressed in all lineages of hematopoietic cells except for T cells and plasma cells.9,10 The BTK mutational consequences affect the expression of hundreds of genes, proteins, and molecular regulators.23–25
The first case reported by Ogden Bruton (1952)26 was an 8-year-old boy who had suffered recurrent invasive pneumococcal infections from the age of 4.5 years and in whom agammaglobulinemia was confirmed by using electrophoresis.26,27 Bruton not only realized that there was a connection between the clinical and absence of the gammaglobulin fraction, but also successfully treated this boy with replacement immunoglobulin therapy.26 Subsequent cases revealed an X-linked pedigree27,28 with family history in 40% of the affected individuals.7,8 Nearly 927 mutations have been associated with the disease in 1757 patients.29 Our group has been the first to publish the molecular diagnosis of XLA in Brazilian patients.30,31 Although it has not been possible to correlate the genetic defect with the severity of the clinical phenotype,7,10,16 it is possible that the nonsense mutations directing to RNA decay may affect BTK expression with its hypoexpression.32
XLA diagnosis evaluationDiagnosing relies first on detailed family history and infection history, with age of onset, frequency and duration of treatments, and if known, organisms that might suggest a primary B-cell defect or a combined B and T-cell immune defect. XLA diagnosis accounts for male patients with onset of recurrent infections most often during the first year of life, when transferred maternal IgG has been catabolized and the mother stops breastfeeding.33 Before the age of 5 years, the child required one or more hospitalizations. In a large study with 783 XLA patients,8 they reported vaccine-associated paralytic poliomyelitis in countries with attenuated live polio vaccination; acute and chronic lung diseases accounted for 41% of the deaths; inflammatory bowel disease and enteroviral meningoencephalitis were more prevalent in South American patients; large granular lymphocyte disease, among others, was enumerated in 20.3% of patients.
Initial laboratory tests include a complete blood count, quantitative serum levels of IgM IgG, IgA, IgE and antibody titers in response to vaccines (tetanus and diphtheria toxoids, Haemophilus influenzae, measles, mumps, rubella, varicella, pneumococcus, hepatitis A or B, influenza virus) to fully validate the immune defect.5,7,8,10,33 The next step is a lymphocyte phenotyping using flow cytometry, which documents normal T-cell numbers but <1–2% B cells. To confirm the diagnosis, genetic testing to look for variants in Btk gene can be performed. A confirmed family history of XLA can serve as a surrogate for genetic testing.[7,33]
Spectrum of bacterial infections in XLASepsis, meningitis, or cellulitis due to H. influenzae and S. pneumoniae in a vaccinated child should elicit an evaluation for PAD.5,7,8,18 After the onset of IRT, serious infections decrease, but they continue to have otitis, sinusitis, and or conjunctivitis.7,18Pseudomonas or Staphylococcal sepsis may occur in infants with neutropenia.34Non sporulating gram negative rod campylobacter jejuni infections cause sepsis, gastrointestinal disease, erysipelas-like skin lesions, pericarditis, and recurrent fever.18 Chronic diarrhea is caused by C. jejuni. Pneumocystis jirovecii pneumonia is rare.18 Fastidious organisms related to C. jejuni or Flexispira rapini have been reported in patients on Ig therapy.18 Systemic Ureaplasma urealyticum or Mycoplasma species are frequent in the respiratory tract, joints and urogenital tract.18
Spectrum of viral Infection in XLASARS-CoV-2 infectionA report of two XLA patients with SARS-CoV-2 infection, aged 34 and 26 years, who developed interstitial pneumonia and lymphopenia, fever, cough, and anorexia, elevation of CRP and ferritin, but never required oxygen ventilation or intensive care.35 Two other adults, XLA and ARA, both with COVID-19, presented with mild symptoms, short duration, no need for drugs, and had a favorable outcome.36 Apart from that, the COVID-19 course was severe in 5 patients affected with CVID, requiring drug treatment and mechanical ventilation.36 It is speculated that the absence of BTK could advantage XLA patients.35 Other three XLA patients (10 y.o., 24 y.o., and 40 y.o.) with COVID-19 displayed strong pro-inflammatory responses in the absence of B cell, failed supportive treatment but recovered after receiving convalescent plasma.37 Therefore, more observational studies are necessary to extend these experiences to other patients with similar genetic immune defects.
EnterovirusXLA patients are susceptible to norovirus and severe enterovirus infections such as Echo virus and vaccine-associated paralytic poliomyelitis (VAPP) from live-attenuated oral poliovirus vaccine (OPV) and Rhinovirus.8,10,24,38 They are possibly also suscetible to arbovirus. However, they are not susceptible to herpes family, cytomegalovirus, varicella, or Epstein Barr virus, and H1N1 virus.24 The use of IRT has decreased the incidence of enteroviral infections manifesting as chronic meningoencephalitis, hepatitis, or dermatomyositis-like infections.21 Early literature on XLA found 15% to 20% of patients infected by enteroviral meningoencephalitis even in the presence of Ig therapy.22 Brain biopsy is recommended as soon as a neurological complication is suspected. Gait disturbances, progressive spastic tetraparesis, intellectual and speech deterioration, no infectious agent identified, periventricular demyelinating and cortical atrophy associated with stereotactic brain biopsy are all signs of progressive multifocal leukoencephalopathy (PML) in agammaglobulinemia.39
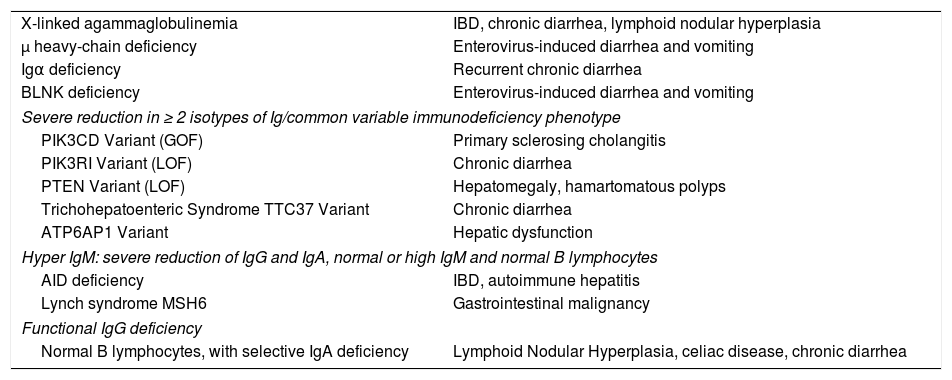
Gastrointestinal manifestationsImpaired humoral immunity and intestinal abnormalities (Table 1) have been observed in XLA, ARA, CVID, IgAD and other genetic defects. Causes of chronic or recurrent diarrhea in XLA and ARA have included Giardia lamblia, rotavirus, Campylobacter fetus, enterovirus, Salmonella, and cryptosporidia.5,10,18 Since gastrointestinal manifestations are present in up to 35% of XLA patients, with IBD/enteritis diagnosed in up to 10% of patients, a close monitoring is necessary.[40]
Predominantly antibody deficiency associated with gastrointestinal manifestations.
| X-linked agammaglobulinemia | IBD, chronic diarrhea, lymphoid nodular hyperplasia |
| μ heavy-chain deficiency | Enterovirus-induced diarrhea and vomiting |
| Igα deficiency | Recurrent chronic diarrhea |
| BLNK deficiency | Enterovirus-induced diarrhea and vomiting |
| Severe reduction in ≥ 2 isotypes of Ig/common variable immunodeficiency phenotype | |
| PIK3CD Variant (GOF) | Primary sclerosing cholangitis |
| PIK3RI Variant (LOF) | Chronic diarrhea |
| PTEN Variant (LOF) | Hepatomegaly, hamartomatous polyps |
| Trichohepatoenteric Syndrome TTC37 Variant | Chronic diarrhea |
| ATP6AP1 Variant | Hepatic dysfunction |
| Hyper IgM: severe reduction of IgG and IgA, normal or high IgM and normal B lymphocytes | |
| AID deficiency | IBD, autoimmune hepatitis |
| Lynch syndrome MSH6 | Gastrointestinal malignancy |
| Functional IgG deficiency | |
| Normal B lymphocytes, with selective IgA deficiency | Lymphoid Nodular Hyperplasia, celiac disease, chronic diarrhea |
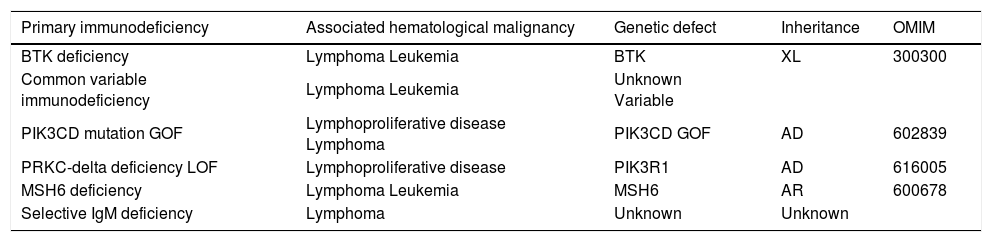
Rates of malignancy in XLA are reported to be between 1.5 and 6%, with patients most likely to develop lymphoproliferative disorders (B or T-cell lymphoma, Hodgkin's lymphoma, myelodysplastic syndrome, leukemia, carcinoma; and, rarely, melanoma, medulloblastoma, neuroblastoma, or gastric cancer and colorectal cancer) (Table 2).7,8,41
Predominantly antibody deficiencies associated with hematological malignancies.
| Primary immunodeficiency | Associated hematological malignancy | Genetic defect | Inheritance | OMIM |
|---|---|---|---|---|
| BTK deficiency | Lymphoma Leukemia | BTK | XL | 300300 |
| Common variable immunodeficiency | Lymphoma Leukemia | Unknown Variable | ||
| PIK3CD mutation GOF | Lymphoproliferative disease Lymphoma | PIK3CD GOF | AD | 602839 |
| PRKC-delta deficiency LOF | Lymphoproliferative disease | PIK3R1 | AD | 616005 |
| MSH6 deficiency | Lymphoma Leukemia | MSH6 | AR | 600678 |
| Selective IgM deficiency | Lymphoma | Unknown | Unknown |
AR, autosomal recessive; AD, autosomal dominant; XL, X-linked; GOF, gain of function.
Arthritis, inflammatory bowel disease (IBD) or other inflammatory conditions have been showed to be prevalent in these patients.5,7,8,10,42 Data from the USIDNET registry showed 12% of XLA patients reporting arthralgia or joint swelling, with 16% having a diagnosis of arthritis, which frequently responds to IRT. Because of lack of immunoglobulins, autoantibody analysis is of no value, and ESR is normal.
Autosomal recessive agammaglobulinemia (ARA)ARA corresponds to 15% of all patients with a B-lymphocyte differentiation arrest, caused by defects of the BCR structure itself, including the μ heavy chain gene mutations (IGHM OMIM 147020), surrogate light chains VpreB and λ5 (IGLL1 OMIM 146770), Igα (CD79A OMIM 112205), Igβ (CD79B OMIM 147245), B-cell linker (BLNK MIM 604515), p110δ (PIK3CD OMIM 602839), p85 (PIK3R1 615214), E47 transcription factor (TCF3 OMIM AD 616941; OMIM AR 147141), SLC39A7(ZIP7) SLC39A7 OMIM 601416), Hoffman syndrome/TOP2B (TOP2B OMIM 126431).1,10,18 Clinically, μ heavy chain gene mutations (IGHM OMIM 147020) is a more severe phenotype, with higher incidence of enteroviral infections, Pseudomonas sepsis, and neutropenia than patients with mutations in BTK. Gene defects of Igα and Igβ are very rare, and the patients have recurrent sinopulmonary infections, chronic diarrhea with malabsorption and dermatomyositis-like manifestations, and sometimes neutropenia. E47 Transcription Factor/TCF3 deficiency autosomal recessive (OMIM: 147141) includes severe, recurrent bacterial infections and failure to thrive, agammaglobulinemia and B-cells have increased expression of CD19+ but absence of a BCR.[1,5,10,18]
Autosomal dominant agammaglobulinemia was reported as a result of mutations in the LRRC8A gene on chromosome 9q34 in a 17-year-old girl (OMIM: 613506) and TCF3 gene on 19p13.3 (OMIM: 616941).8,18
ManagementThe measures to prevent infections are frequent handwashing, good respiratory hygiene, and only ingesting drinking water. IRT has reduced the rate of infections and hospitalizations. However, a few uncommon pathogens to which the donor pool has not been exposed are not precluded. An emerging route of administration is via subcutaneous injections (SCIG); only IgG is replaced. In addition to IVIG, treatment with antibiotics for active infections should be done.7,8,10,18 Regular monitoring is recommended for recurrent pneumonia, bronchiectasis, chronic sinusitis, and chronic bronchitis using imaging studies. Hematopoietic stem cell transplantation (HSCT) is an alternative. The risks of allogeneic HSCT, such as rejection and graft-versus-host-disease, make the treatment option less safe.7 However, gene therapy is emerging as a promising possibility of lifetime cure for these patients, foregoing the need for long-term immunoglobulin therapy, eliminating the risks brought on by recurrent infections, restoring B-lymphocyte functions and potentially ensuring adequate BTK expression on other immune cells.[7]
Severe reduction in at least 2 serum immunoglobulin isotypes with normal or low number of B cells, CVID phenotypeCVID was first recognized in 1954 and has been the most prevalent form of PAD.43 CVID can be described as a collection of late-onset hypogammaglobulinemia syndromes. The diagnosis is commonly applied to male or female patients, age greater than 4 years, with low levels of serum IgG, IgA, and/or IgM, as well as failure to mount antibodies against vaccination, to both protein and carbohydrate antigens.5,6,33
Causes of primary or secondary hypogammaglobulinemia, such as bone marrow infiltrative disease, medications (Azathioprine, Cyclosporine, D-penicillamine, Gold, Sulfasalazine, Carbamazepine, Levetiracetam, Oxcarbazepine, Phenytoin), and immune globulin losses (in the gut or urine), as well as all other genetic defects must be excluded.5,6,17,33 CVID patients have low to normal numbers of circulating B cell, failure in the differentiation of B cells into immunoglobulin-secreting plasma cells, reduced numbers of isotype switched CD27+ memory B cells, increases in CD21 low or increased transitional B cells, reduced T cell numbers, cytokine defects, defective lymphocyte proliferative to mitogens and antigens, abnormal lymphocyte trafficking, dysregulated cellular responses to chemokines, defective dendritic cell and innate immune interactions, uncontrolled T-cell polarization, and more recently described, an inflammatory role of innate lymphoid cells.6,10,11,33 The identification of CVID familial cohorts and the progression of some IgA-deficient patients to CVID indicate a strong contribution of genetics. However, the genetic basis has been determined in only 10% of CVID cases.11,17 Polymorphisms in the costimulatory molecules CD18, CD19, CD20, CD21, ICOS, TACI, and BAFF have all been linked to CVID. This correlation is because B cells from patients with CVID are inhibited in their ability to develop into class-switched memory and plasma cells, a maturation process that requires adequate costimulation. Around 40 genetic defects have been identified in 571 patients with a CVID phenotype from three cohorts: US, Sweden, and Iran; 235 (74 subjects with mutations; 31%), 128 (46 subjects with mutations; 36%), and 208 (113 subjects with mutations; 54%) patients respectively.11 These data show a multiplicity of genes identified, reflecting the complex requirements of B-cell antigen signaling, activation, survival, migration, maturation, and maintenance of antibody-secreting memory B-cell populations to the plasma cell stage.11 Specific clinical patterns of illnesses were also not linked to any given gene defect as there was considerable overlap in clinical presentations.11,17 Chapel et al. (2008)12 identified five distinct clinical phenotypes in CVID: no complications, autoimmunity, polyclonal lymphocytic infiltration, enteropathy, and lymphoid malignancy.12 Currently, the clinical spectrum of CVID consists of mainly two phenotypes: one predominated by recurrent infections, while in approximately 25–50% of patients autoimmune and/or inflammatory features are present, including enteropathy (Table 1), noninfectious immune-mediated lung disease and/or granulomatous disease, which lead to significant morbidity and mortality.10,11,17 Bronchiectasis is frequent, and liver dysfunction with nodular regenerative hyperplasia leading to portal hypertension, or primary biliary cirrhosis and/or granulomatous disease have been reported in 10% of patients. Approximately 25% of patients have splenomegaly and/or generalized lymphadenopathy, leading to concern for malignancies,6,17 particularly non Hodgkin's lymphoma (Table 2), and have an estimated 1.8- to 5-fold increased risk of developing cancers of all types.6,17 For the US and Swedish cohorts, CVID subjects with lymphoid infiltrations or inflammatory or autoimmune complications were more likely to have an identifiable gene, but in both cohorts the majority of subjects CVID with no gene defect specified had no potential gene that could currently be assigned.11
Respiratory manifestationPneumonia caused by H. influenzae and S. pneumoniae may lead to pleurisy, empyema, and bronchospasm, which lead to chronic lung disease, as reported in 28.5% of 473 patients with CVID.6,10,17,44 The survival was significantly reduced compared to those without chronic lung disease. Early prophylactic antibiotics and immunoglobulin replacement therapy (IRT) can reduce respiratory infections and decrease morbidity and mortality from pulmonary disease in PADs.6,10,17,44 Chronic lung disease progresses in many patients, in part by immune dysregulation occurring independent of infection and also due to mucosal IgM and IgA responses’ deficiencies. Chest imaging and pulmonary function testing (PFT) form the basis of the diagnostic work-up of chronic lung disease, with lung tissue biopsy when indicated.44
Asthma and chronic obstructive pulmonary disease (COPD) has been reported in IgAD, 15% to 50% of those with CVID and 5% of patients with XLA. The etiologies must be considered in PADs to prevent it from being misdiagnosed as asthma.6,17,44
Bronchiectasis is associated with a history of pneumonia, older age, chronic cough with purulent sputum, occasional hemoptysis, and dyspnea, as well as reduced levels of CD4+ T cells and lower levels of IgA and/or IgM. Airflow obstruction is often evident on PFT, and diagnosis is made by CT.17,44
Interstitial lung disease (ILD) occurs in 10–20% of patients with CVID, but 64% of patients with respiratory symptoms have images of ground glass opacity and/or numerous pulmonary nodules in the CT. ILD is more common in CVID associated with autoimmunity and T-cell deficiency than in XLA, and in IgAD patients with autoimmunity and IgG subclass deficiency. ILD pathology in patients with PAD consists in forms of benign pulmonary lymphoproliferation, usually follicular bronchiolitis (benign hyperplasia of lymphoid follicles), lymphocytic interstitial pneumonia (LIP), or nodular lymphoid hyperplasia.6,17,44
Granulomatous inflammation and organizing pneumonia may also be found in the lungs of patients with PAD together with one of the lymphoproliferative pathologies, and the term granulomatous and lymphocytic interstitial lung disease (GLILD) is frequently used to describe the ILD associated with PAD.6,17,44 The role of B cells in determining lung inflammatory disorders is also demonstrated by the observation that GLILD occurs in 10% of patients with CVID and can be treated with B-cell-depleting drugs.44
Neurologic manifestationsMeningitis is the most common neurologic manifestation of CVID. The rare neurological diseases in CVID include axonal sensory-motor polyneuropathy as autoimmune involvements, transverse myelitis, progressive neurodegeneration, cerebral vasculitis causing recurrent occipital headaches, brain granulomatous lesions, free radical mediated neuronal damage due to vitamin E deficiency, including sensory loss, ataxia, retinitis pigmentosa, subacute combined degeneration of the cord secondary to vitamin B12 deficiency, peroneal muscular atrophy, Guillain-Barré syndrome and myasthenia gravis.6,10,11,17,45
Severe reduction in serum IgG and IgA with normal/elevated IgM and normal numbers of B cells, hyper IgMHyper IgM syndrome“Hyper-IgM syndromes (HIGM)” are rare, characterized by impaired production of switched immunoglobulin isotypes (IgG, IgA, IgE) and normal or elevated IgM levels. Some deficiencies are caused by defects in CSR machinery and are intrinsic B-cell defects, due to variants in AID and UNG.5,10,46 In contrast, CD40 ligand (CD40L) and CD40 deficiencies are combined immune defects with impaired interaction between activated CD4+ T cells expressing CD40L and cell types expressing CD40, which include B cells, dendritic cells, monocytes/macrophages, platelets, and activated endothelial/epithelial cells.5,10,46 CD40L on activated T cells and cognate interactions with CD40 on B cells result in B-cell proliferation, adhesion, and finally, differentiation.5,10 CD40L deficiency is inherited as an X-linked trait and is the most common form of HIGM; its estimated frequency is 2:1,000,000 males.10 CD40L deficiency often presents in infancy with recurrent sinopulmonary infections, caused by Streptococcus pneumoniae and Haemophilus influenzae, and higher risk of opportunistic infections by Pneumocystis, Cryptosporidium, and Histoplasma organisms. Patients can often have chronic and recurrent diarrhea due to infection with Cryptosporidium parvum, with increased risk of biliary tract diseases such as sclerosing cholangitis and cholangiocarcinoma.5,10,46 Chronic viral hepatitis, CMV infections, Cryptococcus and Toxoplasma infections, JC virus-related enteroviral meningoencephalitis and progressive multifocal leukoencephalopathy (PML)5,10,46 have been reported. Recurrent oral ulcers and proctitis, often associated with chronic or cyclic neutropenia are common in half of the patients.10 Autoimmune complications include thrombocytopenia and autoimmune hemolytic anemia.5,10 Patients with CD40L deficiency also have an increased risk of malignancies of hepatobiliary origin including hepatocellular carcinoma, cholangiocarcinoma, peripheral neuroectodermal tumors of the gastrointestinal tract and with a lesser incidence of lymphoma.5,10 CD40 recessive mutations in the B-cell surface receptor CD40 (OMIM: 109535) are very rare, and the patients have similar clinical features as CD40L deficiency.10
Isotype, light chain, or functional deficiencies with generally normal numbers of B cellsSelective IgA deficiencySelective IgA deficiency (SIGAD) is the most common PAD, with incidence varying between 1:143 and 1:18,500.5,10 It affects males and females equally and is defined as a serum IgA level of less than 7mg/dL and normal levels of serum IgG and IgM in a patient older than 4 years.5,10,33 Primary IgA deficiency must be distinguished from secondary causes due to anticonvulsant medications (phenytoin, carbamazepine, valproic acid), anti-rheumatic drugs (sulfasalazine, hydroxychloroquine), and nonsteroidal anti-inflammatory drugs.10,46 Impaired switching to IgA or a failure of IgA producing B-cell maturation into IgA secreting plasma cells have been intensively investigated.5,10 Clinically, two-thirds of patients remain asymptomatic, whereas symptomatic patients suffer from allergies, recurrent sinopulmonary and mucosal infections, both infectious and non-infectious gastrointestinal diseases, and gastrointestinal and lymphoid malignancies.5,10 Autoimmunity also appears. The pathophysiology of SIGAD remains unknown.10 Associations with selected MHC alleles and higher frequency within families with autoimmunity or other immune defects have been reported. Patients with SIGAD have progressed to CVID,10 suggesting further monitoring.
Transient hypogammaglobulinemia of infancy (THI)A disease first described in 1956,47 the patients with THI have low IgG levels (2 SD below the mean for age-match controls) with possible involvement of IgA and, less frequently, IgM that spontaneously return to normal, usually within 2–3 years of age, although it often varies.5,10 Most subjects with THI remain asymptomatic; however, in some patients it can be associated with a higher rate of recurrent infections.5,10,47 A prospective study of infants with THI showed that those with a low number of memory B cells and inability to produce IgG in vitro were associated with persistence of hypogammaglobulinemia and an increased risk of infection beyond 2 years of age.10 The evaluation of THI should include specific antibody responses to age-appropriate vaccines and immunophenotyping by flow cytometry to exclude other immune defects.5,10 Vaccines’ antibody responses are often intact.5,10 THI is self-limited, and patients should be monitored over time until levels have normalized.5,10 However, the defect may be a harbinger of a more permanent immune defect.[10]
ConclusionThe IUIS expert committee assembled these complex experiments on the nature of PADs, using only two biomarkers: the serum Ig levels and B-cell number. The physiopathology of these disorders involves variants in cytoplasmic tyrosine kinase BTK in XLA, and variants of the components of the BCR in ARA. Both reveal the secrets of BCR signals for the maintenance of mature B-cell populations. CVID revealed that BAFF, CD19, CD20, CD81, and CD21 receptors are essential for amplification of BCR signaling. The X-linked HIGM syndrome revealed that CD40L is essential for Ig class switch, germinal center formation, and CD27+ memory B cells. Other HIGM defects, CD40, AID and UNG deficiency, PIK3R1 and INO80 chromatin remodeling complex illustrate the additional requirements for these functions. These advances suggested important avenues for therapies. The advent of next-generation sequencing has greatly facilitated the search for novel causative genes in PADs, mainly in CVID. Currently, the easy access and low cost of blood count and immunoglobulin dosage justify that they be performed in the second half of the normal child's life, as an assessment of the ontogeny of the immune system.
FundingThis work was supported by grant no. #2016/25615-6, São Paulo Research Foundation (FAPESP).
Conflicts of interestThe author declares no conflicts of interest.




