

Congenital hyperinsulinism (CHI) is a heterogeneous genetic disease characterized by increased insulin secretion and causes persistent hypoglycemia in neonates and infants due to dysregulation of insulin secretion by pancreatic β cells. Babies with severe hypoglycemia and for whom medical treatment has been ineffective usually require surgical treatment with near-total pancreatectomy. To evaluate the clinical and surgical aspects affecting survival outcomes in babies diagnosed with CHI in a single tertiary care center.
MethodsRetrospective Cohort study involving a single university tertiary center for the treatment of CHI. The authors study the demographics, clinical, laboratory, and surgical outcomes of this casuistic.
Results61 % were female, 39 % male, Birth weight: 3576 g (±313); Age of onset of symptoms: from the 2nd hour of life to 28 days; Time between diagnosis and surgery ranged between 10 and 60 days; Medical clinical treatment, all patients received glucose solution with a continuous glucose infusion and diazoxide. 81 % of the patients used corticosteroids, 77 %. thiazide, 72 % octreotide, 27 % nifedipine; Neurological sequelae during development and growth: 54 % had some degree of delay in neuropsychomotor development, 27 % obesity. Surgery was performed open in 6 and 12 minimally invasive surgery (MIS). Histopathology: 2 focal and 16 diffuse, Length of stay (days) was lower in MIS (p < 0.05). Survival was 100 %.
ConclusionsCHI is a rare and difficult-to-manage tumor that must be performed in a multidisciplinary and tertiary center. Most surgical results are good and the laparoscopic approach to disease has been the best choice for patients.
Congenital hyperinsulinism (CHI) is a heterogeneous genetic disease characterized by increased insulin secretion and causes persistent hypoglycemia in neonates and infants due to dysregulation of insulin secretion by pancreatic β cells.1,2 Histopathologic forms of CHI can have diffuse or focal involvement and are predominantly caused by inactivating mutations of ABCC8 or KCNJ11, the two genes that encode the β-cell ATP-dependent potassium channel (KATP).3 The incidence of CHI is 1 in every 40,000 to 50,000 live births in the general population, and paternal consanguinity can reach 1 in 2500 live births.4
CHI is a neonatal clinical emergency because the early diagnosis and management of hypoglycemia is very important to prevent permanent brain damage such as cerebral palsy, epilepsy, and death.5,6 The diagnosis is basically made by measuring and testing blood glucose, insulin, and glucagon. In normal children, when the blood glucose concentration falls below 60 mg/dL (3.3 mmol/L), inhibition of insulin secretion occurs. However, in the CHI this suppression does not happen with insulin secretion independent of glycemic levels, leading to severe hypoglycemia.7
Clinical treatment consists of frequent oral feeding, oral feeding via nasogastric tube, or, more rarely, gastrostomy. For infants who are fed, and it is not possible to reach normoglycemia, there is a need for intravenous infusion of dextrose.8 Children with severe hypoglycemia and for whom medical treatment has been ineffective usually require surgical treatment with near total pancreatectomy.9 Positron emission tomography ± Computed tomography (PET/CT) imaging with 18-fluoroDOPA can help localize lesions and allow partial pancreatectomy with cure in almost all patients with focal CHI with surgical treatment performed by laparotomy or minimally invasive surgery approach (MIS).10,11
There are few reference centers for the complete treatment of CHI, so the authors’ objective was to review the clinical and surgical characteristics of a tertiary CHI treatment center in Brazil.
MethodsPatient population and data collectionThe inclusion criteria were all babies with CHI confirmed by clinical and laboratory tests and histopathological surgical findings submitted to surgery in the studied institution. Exclusion criteria were children with any hypoglycemia acquired. Data were taken from medical records and the following data were analyzed: a) Demographics: origin, gender, birth weight, age of the symptoms, age of diagnosis, the time between diagnosis and surgery, type of medical clinical treatment used, resolution of hypoglycemia, neurological sequelae during development and growth; b) Laboratory tests: glycemia, insulinemia, C-peptide, blood gas test, ammonia, lactate, growth Hormone (GH), cortisol, Adrenocorticotropic Hormone (ACTH) and c) Surgery: kind of procedure (open vs MIS), kind of surgery (total or partial), age of surgery, weight in surgery, The American Society of Anesthesiologists Classification Risks (ASA), time of anesthesia, complications of anesthesia, time of surgery, complications of surgery, reoperation, additional surgery, tumor classification (diffuse or focal), genetics investigation, survival and follow up. All 18 patients were diagnosed with PET scan.
Statistical analysisData were compared according to the following outcomes: Comparison between open versus MIS. The authors determined the type of data distribution using the Kolmogorov–Smirnov normality test. Continuous numerical variables showing a nonparametric distribution were evaluated using the Mann–Whitney test. The GraphPad Prism program (GraphPad Prism 8.0.1; GraphPad Software Inc., San Diego, CA, USA) was used to analyze the data. Values of p < 0.05 were considered statistically significant.
Ethics approval and consentThe authors performed a retrospective and descriptive study of 15 years (from January 2007 to January 2022) involving a Cohort of 18 babies diagnosed with CHI assisted at a single tertiary center in the Clinic Hospital of Ribeirao Preto Medical School – University of Sao Paulo. This study was performed in accordance with the declarations of Helsinki and the project was approved by the clinical research unit of Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da USP – Plataforma Brasil - CAAE: # 11631912.1.0000.5440.
ResultsIn the selected period from January 2007 to January 2023, 18 pancreatectomies were performed due to refractoriness to conservative treatment.
The patients came from 8 different states in Brazil such as São Paulo, Distrito Federal, Espírito Santo, Goiás, Pernambuco, Minas Gerais, Amapá e Rio Grande do Sul. In relation to gender, 11 were female (61 %) and 7 were male (39 %); the average birth weight was 3576 g (±313); the age of onset of symptoms showed that 8 started in the neonatal period, ranging from the 2nd hour of life to 28 days of life. The age of diagnosis ranged between 1 and 28 days; the time between diagnosis and surgery ranged between 10 and 60 days.
In relation to the medical clinical treatment, all patients received glucose solution with a continuous glucose infusion rate ranging from 4 to 18 mg/kg/min and diazoxide at doses from 5.1 to 15 mg/kg/day. Most (81 %) patients used corticosteroids. The association of thiazide was used in 77 % of the cases, with doses of 2 to 5 mg/kg/day. Nifedipine was used in 27 % of patients with doses up to 1.1 mg/kg/day. Octreotide was used in 72 % of the patients, with doses of 5 to 50 mcg/kg/day. Glucagon was used in 4 patients, with bolus doses in 3 of the cases and in 1 of the patients in continuous infusion. Sirolimus was used in 18 % of the patients, with doses up to 1.2 mg/m2/day. Only 2 patients underwent the glucagon response test, with an increase in values above 30 pg/mL between the measurements taken after 30, 60, 90 and 120 min.
Dietary intake was provided by offering diet orally in 9 % of patients to the resolution of hypoglycemia. The diet of the other patients was administered by orogastric tube or by gastrostomy. Other medications were prescribed in the treatment, anti-reflux medication in 36 % and anticonvulsants in 27 % of patients.
All patients were able to normalize their diets in the late postoperative period, allowing them fasting time flexibility that was not possible before surgery. In 45 % of the patients, maintenance of the gastrostomy was necessary to ensure adequate nutritional intake.
All patients evolved to resolution, with no deaths recorded. In the follow-up, it was found that 54 % of the patients had some degree of delay in neuropsychomotor development, 27 % of the patients evolved Diabetes Mellitus, and 18 % with obesity. Lately, 27 % had height above 3 standard deviations for age and 1 patient had early adrenarche.
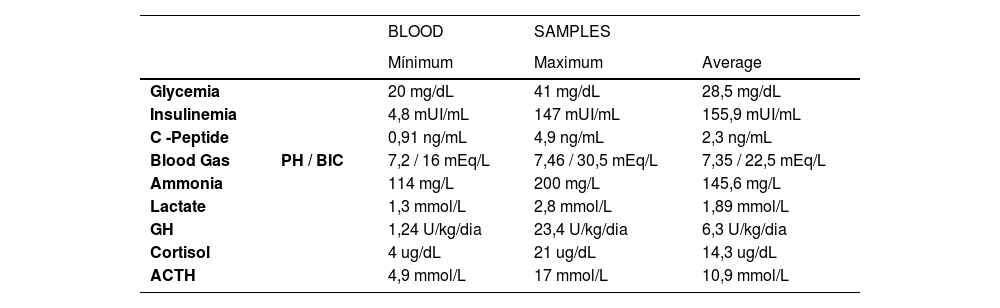
The results of the laboratory tests for diagnosing CHI are in Table 1.
Results of critical blood samples.
In relation to the anesthetics risk, the ASA I and II (low risk) were classified in 12 and 6 patients respectively. There were no complications during the anesthesia procedure. Median age at surgery was 86 days (range 59 to 100 days).
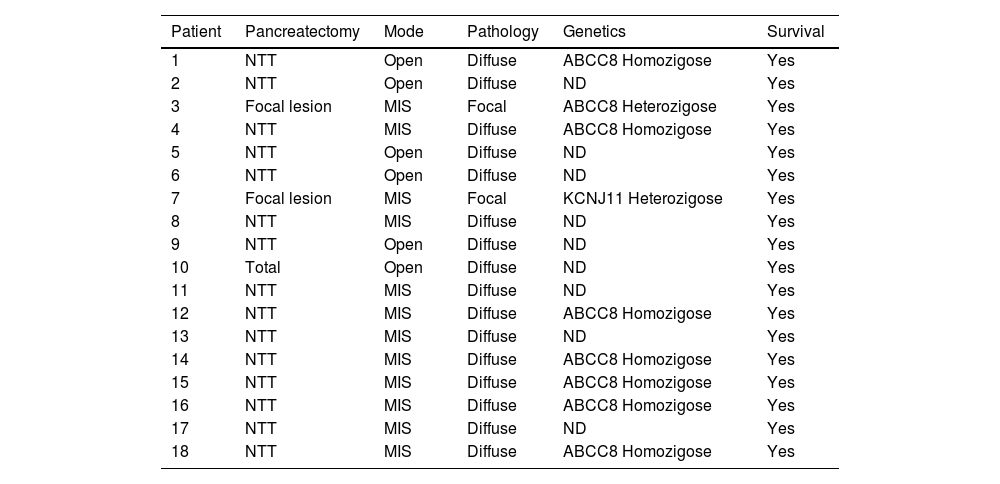
Near to total pancreatectomy was performed in 16 patients, total pancreatectomy in only 1 patient, and the last one was performed with pancreatic remnants ranging from 1 to 15 %. Of the 18 surgeries described, 6 of them (33 %) were performed by an open approach, one of them after conversion of an MIS, and the remaining 12 were by MIS. In the anatomopathological classification, 2 patients had focal disease and the remaining 16 had diffuse disease. The genetic study confirmed mutations in the ABCC8 and KCNJ11 genes in 50 % of the operated patients. The finding of a mutation in heterozygosity of paternal origin in the ABCC8 gene occurred in the 2 patients diagnosed by CT-Pet Scan as focal before surgery, in one patient the focal lesion was in the tail, and in the other, the lesion was in the body of the pancreas. All cases in which he identified a homozygous mutation had diffuse disease confirmed by histological evaluation. All patients evolved to resolution and without deaths (Table 2).
Characteristics of surgical treatment, pathology findings and the genetic investigation from 18 patients with CHI.
NTT, Near to total, MIS: Minimal Invasive Surgery; ND, Not determinate.
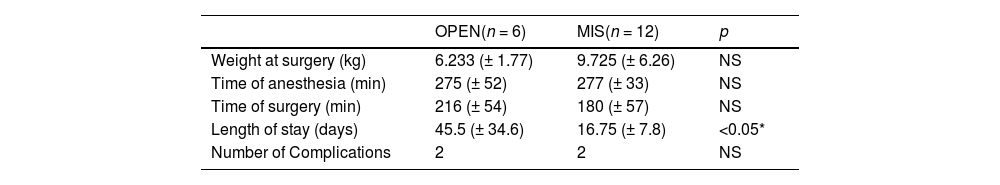
A total of major complications was considered until 15 days post-operative. In 3 patients, a second surgical approach was necessary to remove more pancreatic tissue due to the persistence of hypoglycemia in the late postoperative period (2 open and 1 MIS), 1 patient had Y-Roux additional with duodenal damage after MIS dissection. The comparison between open versus MIS is set in Table 3.
Comparison between open versus MIS.
| OPEN(n = 6) | MIS(n = 12) | p | |
|---|---|---|---|
| Weight at surgery (kg) | 6.233 (± 1.77) | 9.725 (± 6.26) | NS |
| Time of anesthesia (min) | 275 (± 52) | 277 (± 33) | NS |
| Time of surgery (min) | 216 (± 54) | 180 (± 57) | NS |
| Length of stay (days) | 45.5 (± 34.6) | 16.75 (± 7.8) | <0.05* |
| Number of Complications | 2 | 2 | NS |
The clinical complications and follow-up after the surgery are represented in Figure 1.
Discussion
Infants with persistent CHI should be evaluated within a strong network of diagnostic, treatment, and research institutes.12 The age of onset of hypoglycemia reflects the severity of CHI. Early diagnosis and appropriate treatment are important for favorable mental outcomes.13
Imaging the pancreas in infants and children is quite challenging in CHI and the 18F-FDOPA has been used quite successfully in diagnosis because PET scan plays an important role in distinguishing between focal and diffuse forms of the disease since anatomical imaging alone, including ultrasound, CT scan, and magnetic resonance imaging (MRI), are rarely able to detect any abnormality in the pancreas. PET also helps guide surgery by providing information about the location of focal cells in hyper-functioning islets.14
All the studied patients had the diagnosis performed by PET scan, which was fundamental for the best surgical result. Many of the patients came from different states of the country, demonstrating the importance of a multidisciplinary CHI care center. Most were in the neonatal period. Of the 18 medical records reviewed, it was found that 94 % (17/18) of the patients had the onset of symptoms between the first week and 28 days of life, showing that the disease appears predominantly in the neonatal period. Diffuse disease was also found in 89 % (16/18) of the present sample. These data are higher than those reported in other studies that point to a 60 % prevalence of diffuse disease in CHI undergoing surgery.15
In a series involving 500 patients with CHI who underwent pancreatectomy, they found that 45 % of them had diffuse disease and 55 % had focal disease.11
In the present study, all patients received diazoxide as a drug treatment, with dosages ranging from 4.1 to 15 mg/kg/day. Diazoxide is the first-line treatment indicated for patients with CHI, however, it is a costly drug for the country's health system.
In all cases operated at the studied center, genetic investigation was carried out with a predominance of focal disease in 16 patients, and only 2 patients were identified with a paternal heterozygous mutation. The authors were unable to justify the finding of a higher predominance of diffuse disease.
Regarding the results of the genetic study, in almost half of the samples, no changes were found in the 11 genes studied. Regarding the results of the genetic study, in almost half of the sample, no changes were found in the 11 genes studied. Of the 18 patients, 9 genes with mutations were identified, respectively 8 in the ABCC8 gene and 1 in the KCNJ11 gene.
These data agree with the other authors who point out that mutations in the KCNJ11 and ABCC8 genes are responsible for approximately 40 to 50 % of CHI.16 Other authors found 55 % of patients with genetic alterations, with mutations in the ABCC8 gene and 12 % in the KCNJ11 gene being 71 % of that total.17
Open surgery is the classic approach to pancreatic resection in CHI. However, recent advances in laparoscopic surgery have led to pancreatectomy near to total being performed for diffuse forms and distal pancreatectomy for focal lesions distal to the head of the pancreas.18
The amount of resected pancreas was progressively achieved, initially resecting 75 % and later 95 %.19 However, after two decades of 95 % pancreatectomy, there is still a high incidence rate of diabetes with increasing age, developing in 69 % of children followed for more than four years. The 95 % failure of pancreatectomy to prevent hypoglycemia in one-third of children with CHI and the ultimate development of diabetes in at least two-thirds indicates that an alternative treatment strategy has not yet been resolved.20
Casuistic of surgical treatment of CHI and pancreatic tumors are restricted to a few centers in the world and the number of patients above 15 is considered high.21 Some tertiary centers have hundreds of cases of CHI focused on multidisciplinary treatment.11,22-25
Laparoscopic resection can be considered a safe and effective treatment with minimal morbidity and excellent results for most pediatric pancreatic tumors. A study carried out to evaluate the feasibility, safety, and efficacy of the surgical treatment of CHI comparing eight patients submitted to open pancreatectomy versus ten laparoscopic for CHI showed a safe and significantly faster feeding time for the laparoscopic route.26
The initial results show that it is safe and the level of complication equal to that of the open procedure, but with the advantage of a shorter hospital stay in the laparoscopic approach. Performing pancreatectomy proved to be effective in the treatment of CHI with a resolution rate of 100 % of patients.
One-third of children with CHI have feeding problems of which 93 % require anti-reflux medication, and 75 % require nasogastric tube feeding and gastrostomy. These findings are directly related to the persistence of hyperinsulinism.27 Among the postoperative complications, exocrine pancreatic insufficiency (54 %), delay in neuropsychomotor development (54 %), need for gastrostomy (45 %), and maintenance of hypoglycemia in the immediate postoperative period (36 %) stand out, tall stature (27 %), diabetes mellitus (27 %), and obesity (18 %).
The rate of post-surgical hypoglycemia for patients with diffuse disease ranges from 18 to 49 %.11,28 The present study has the limitation of being a retrospective study with a representative series of cases, with the initial experience of the laparoscopic route for the surgical approach of the disease.
Therefore, this study presented the clinical and laboratory characteristics of patients with CHI who did not respond to drug treatment and who required surgical treatment to resolve recurrent episodes of hypoglycemia. Surgical treatment was successful in 100 % of patients, with MIS being the most appropriate approach for focal or diffuse cases. The treatment of CHI must be performed in a tertiary center and have a multidisciplinary approach with a rapid surgical indication to avoid severe neurological sequelae.




