

To characterize a sample of Brazilian patients with maple syrup urine disease (MSUD) diagnosed between 1992 and 2011.
MethodsIn this retrospective study, patients were identified through a national reference laboratory for the diagnosis of MSUD and through contact with other medical genetics services across Brazil. Data were collected by means of a chart review.
ResultsEighty‐three patients from 75 families were enrolled in the study (median age, 3 years; interquartile range [IQR], 0.57‐7). Median age at onset of symptoms was 10 days (IQR 5‐30), whereas median age at diagnosis was 60 days (IQR 29‐240, p=0.001). Only three (3.6%) patients were diagnosed before the onset of clinical manifestations. A comparison between patients with (n=12) and without (n=71) an early diagnosis shows that early diagnosis is associated with the presence of positive family history and decreased prevalence of clinical manifestations at the time of diagnosis, but not with a better outcome. Overall, 98.8% of patients have some psychomotor or neurodevelopmental delay.
ConclusionIn Brazil, patients with MSUD are usually diagnosed late and exhibit neurological involvement and poor survival even with early diagnosis. We suggest that specific public policies for diagnosis and treatment of MSUD should be developed and implemented in the country.
Caracterizar uma amostra de pacientes brasileiros com a doença da urina de xarope de bordo (DXB) diagnosticados entre 1992 e 2011.
MétodosOs pacientes foram identificados por meio de um laboratório de referência nacional para o diagnóstico de DXB e por meio do contato com outros serviços de genética médica no Brasil. Os dados foram coletados por meio de uma revisão de prontuários.
ResultadosForam incluídos no estudo 83 pacientes de 75 famílias (idade média: três anos; intervalo interquartil (IQR): 0,57‐7). A idade média no surgimento dos sintomas era de 10 dias (IQR: 5‐30), ao passo que a idade média no diagnóstico era de 60 dias (IQR: 29‐240; p=0,001). Somente três (3,6%) pacientes foram diagnosticados antes do surgimento de manifestações clínicas. Uma comparação entre pacientes com (n=12) e sem (n=71) um diagnóstico precoce mostra que o diagnóstico precoce está associado à presença de histórico familiar positivo e à redução na prevalência de manifestações clínicas no momento do diagnóstico, porém sem melhor resultado. Em geral, 98,8% dos pacientes têm algum atraso no desenvolvimento psicomotor ou neurológico.
ConclusãoNo Brasil, os pacientes com DXB normalmente recebem um diagnóstico tardio e exibem um envolvimento neurológico e baixa sobrevivência, mesmo com um diagnóstico precoce. Sugerimos que políticas públicas específicas para o diagnóstico e tratamento da DXB sejam desenvolvidas e implementadas no país.
A doença da urina de xarope de bordo (DXB) é uma disfunção genética recessiva autossômica causada pela deficiência na atividade do complexo alfa‐cetoácido‐desidrogenase de cadeia ramificada (BCKDC). A deficiência desse complexo de enzimas leva a níveis elevados de aminoácidos de cadeia ramificada (BCAA) leucina, valina e isoleucina. A leucina e seu ácido 2‐oxo‐isocapróico cetoanálogo são particularmente tóxicos ao sistema nervoso central (SNC). Apesar de a incidência de DXB ao redor do mundo normalmente ser estimada em 1:185.000 recém‐nascidos (RN),1 os dados obtidos de exames em recém‐nascidos sugerem que essa taxa pode ser maior; na Alemanha, por exemplo, a incidência está estimada em 1:133,000 RN2 e em algumas comunidades menonitas e holandesas na Pensilvânia, Estados Unidos, a taxa pode chegar até 1 a cada 200 nascidos vivos.3
O exame neonatal por meio de espectrometria de massa em tandem (MS/MS), também conhecida como triagem neonatal ampliada, permite o diagnóstico de DXB enquanto o paciente ainda está assintomático, bem como o início do tratamento precoce – dois fatores essenciais na melhoria do quadro clínico.3 Antes da introdução da triagem neonatal ampliada, acreditava‐se que a forma grave (DXB clássica) contabilizava 75‐80% dos casos,4 porém dados recentes sugerem que as formas mais leves de DXB podem contabilizar até 50% dos casos diagnosticados.5 Na forma clássica, os sintomas ocorrem primeiramente entre o 4° e o 7° dia de vida e, normalmente, incluem alterações respiratórias, encefalopatia, um odor característico, convulsões e coma.6 Na fase aguda, é necessário um tratamento imediato e agressivo para reduzir os níveis de leucina, o que deve consistir em uma perfusão com alto teor de glicose para estimular a secreção de insulina e suprimir o catabolismo de proteínas. Caso isso não funcione, poderão ser necessárias intervenções invasivas, como diálise peritoneal, hemodiafiltração ou hemodiálise. Durante a fase de manutenção, o tratamento normalmente consiste na restrição dietética de BCAA e suplementação com tiamina e uma fórmula alimentar sem BCAA,6‐8 apesar de o transplante de fígado ser uma boa opção.9‐11
O Programa Nacional de Triagem Neonatal foi implementado em 2001 e não inclui o exame de DXB. A fórmula alimentar sem BCAA, produto de alto custo, não é fornecida pelo Sistema Único de Saúde (SUS). Adicionalmente, os testes de laboratório necessários para o diagnóstico dessa doença também não são fornecidos pelo SUS e estão disponíveis apenas em alguns centros universitários selecionados ou laboratórios médicos privados. A respeito do transplante de fígado, não existe uma rede nacional coordenada que vise a melhorar as condições dos transplantes de fígado para pacientes com disfunções metabólicas. Por fim, não existem dados sobre a prevalência dessa doença no Brasil.
O objetivo deste estudo é descrever o perfil dos pacientes brasileiros com DXB de 1992 a 2011 de forma a contribuir para a consolidação das políticas públicas específicas para DXB no país.
MétodosEste estudo retrospectivo, multicêntrico e longitudinal foi aprovado pelo Conselho de Revisão Institucional.
Os pacientes foram identificados a partir dos registros do Laboratório de Erros Inatos do Metabolismo do Serviço de Genética Médica, serviço universitário que atua como centro de referência nacional no diagnóstico e tratamento de erros inatos do metabolismo, e a partir dos registros do Serviço de Informações sobre Erros Inatos do Metabolismo (Siem, feitos pelo mesmo Serviço de Genética Médica.12 Esse laboratório provavelmente é responsável pela maioria dos diagnósticos de DXB no país; a propedêutica necessária é feita sem custo ao paciente ou médico solicitante e normalmente é financiada por órgãos financiadores de pesquisa. A quantificação dos BCAAs por cromatografia líquida de alta eficiência (CLAE) e pela análise de ácidos orgânicos na urina está disponível no laboratório desde 1994; a análise automática de aminoácidos e MS/MS também está disponível, porém a detecção de aloisoleucina já não é mais feita. O Siem é um serviço de linha direta gratuito, estabelecido em 2001, que fornece informações a médicos e outros profissionais de saúde envolvidos no diagnóstico e tratamento de pacientes com suspeita de Erros Inatos do Metabolismo (EIMs) ou EIMs confirmados.
Para ser incluído no estudo, um paciente deve apresentar: 1) aumento significativo nos níveis de BCAA no sangue em mais de uma mensuração, conforme determinado por um método padrão‐ouro (quantificação de BCAA com base na CLAE, no analisador automático de aminoácidos ou na MS/MS); e 2) diagnóstico da DXB confirmado bioquimicamente e estabelecido entre 1992 e 2011.
Os formulários de coleta de dados foram preenchidos para cada paciente por seu médico atendente ou por um dos pesquisadores do estudo por meio de uma análise dos registros e gráficos disponíveis do paciente. Para pacientes falecidos, os dados do último registro disponível foram considerados para a data de inclusão no estudo.
Definição das variáveis do estudoFoi considerado diagnóstico “precoce” aquele em pacientes diagnosticados antes do 15° dia de vida. A duração da doença até o diagnóstico foi definida como o tempo decorrido entre o início das manifestações clínicas e o diagnóstico bioquímico da DXB. A presença e a gravidade do atraso psicomotor e no desenvolvimento neurológico foram avaliadas com base nas impressões do neurologista ou pediatra que atendeu cada paciente. A DXB foi classificada em variantes de acordo com os critérios normalmente citados na literatura.1
Análise estatísticaForam feitas análises estatísticas no ambiente de software 18.0 do Statistical Package for the Social Sciences (SPSS®, Statistics for Windows, Chicago, EUA). As variáveis foram levadas em consideração na análise apenas se os dados estivessem disponíveis para no mínimo 60% da amostra.
Na análise descritiva, os dados foram expressos como frequências absolutas e relativas. As variáveis contínuas distribuídas assimetricamente foram expressas como medianas e intervalos interquartis. Foram usados o teste qui‐quadrado e o teste exato de Fisher para determinar associações entre as variáveis categóricas. Foram usados o teste de Kruskal‐Wallis e o teste U de Mann‐Whitney para comparar as medianas de características diferentes. O nível de significância foi estabelecido em 5%.
ResultadosForam identificados 119 pacientes com comprovação clínica ou laboratorial da DXB (“pacientes com possível diagnóstico da DXB”), dos quais 83 atendiam aos critérios de inclusão. Desses, 48 estavam vivos na época do estudo, 20 vieram a óbito antes do início do estudo e 15 não apresentavam informações completas de sobrevida.
Os pacientes envolvidos no estudo vieram de todas as cinco regiões do Brasil. A idade média no momento da inclusão no estudo era de três anos (IQR: 0,57‐7 anos; intervalo: 30 dias‐23 anos). Dos participantes, 46 (55,4%) eram do sexo masculino, 75 (90,4%) não foram relacionados e 14 (18,7%) apresentavam histórico familiar de DXB. Foi relatada consanguinidade em 17 famílias (22,7%).
DiagnósticoA idade média no diagnóstico era de 60 dias (IQR: 29‐240 dias; intervalo: sete dias‐10 anos). O nível médio de leucina no diagnóstico era de 1.693μmol/L (IQR: 965‐2.836μmol/L; faixa de referência: 80‐200μmol/L).
Apresentaram manifestações clínicas de DXB no momento do diagnóstico 80 pacientes (96,4%) (idade média no início dos sintomas: 10 dias; IQR: cinco‐30 dias; intervalo: um dia‐dois anos). As manifestações mais comuns foram convulsões (51,2%) e hipoatividade (50%). Outros sintomas apresentados incluíram alimentação deficiente, sucção fraca e alterações no padrão de respiração (48,7% cada), hipotonia (48,2%), odor característico (42,5%), letargia (41,2%), acidose metabólica (31,2%), vômito (30%) e encefalopatia (20%). O odor característico da DXB foi relatado por prestadores de serviços de saúde como um aroma forte ou doce, “semelhante a molho de soja” ou “semelhante a caramelo”, que era mais detectável em pacientes hospitalizados devido à descompensação metabólica. Havia uma diferença estatisticamente significativa entre a idade média no início dos sintomas e a idade média no diagnóstico (p=0,001).
A figura 1 mostra a distribuição do número de diagnósticos por ano e revela uma tendência crescente em diagnósticos durante o período do estudo. Contudo, a comparação da idade média no diagnóstico entre 1992 e 2001 (90 dias; IQR: 36‐270; n=31) e entre 2002 e 2011 (53 dias; IQR: 20‐202; n=52) não revelou diferenças estatisticamente significativas (p=0,053). O tempo médio decorrido entre o início dos sintomas e o diagnóstico foi de 60 dias (IQR: 28‐240) durante a primeira década do estudo e 37 dias (IQR: 9‐180) durante a segunda década (p=0,075). Considerando todos os pacientes com DXB que estavam vivos em 2011 (n=48), 13 haviam sido diagnosticados entre 1992 e 2001 e 35 entre 2002 e 2011.

Número de diagnósticos de DXB no Brasil e tendência de 1992‐2011.
O Siem é um serviço de linha direta gratuito, estabelecido em 2001, que fornece informações a médicos e outros profissionais de saúde envolvidos no diagnóstico e tratamento de pacientes com suspeita EIMs ou EIMs confirmados.
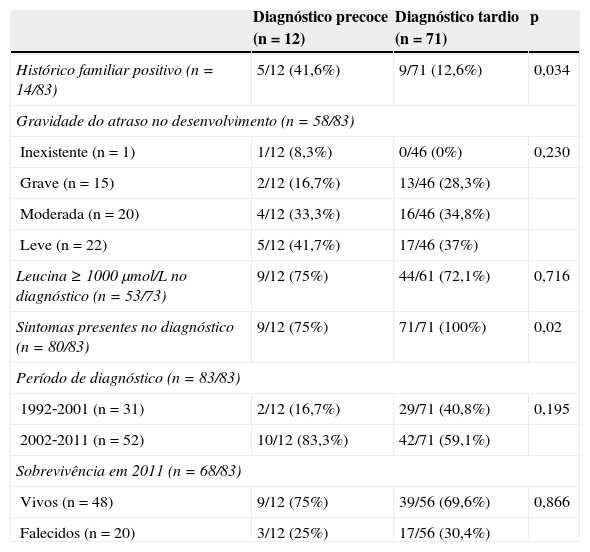
Somente 12 pacientes haviam sido diagnosticados precocemente. Em três desses casos, o diagnóstico foi feito antes do início dos sintomas como resultado de uma triagem neonatal em um laboratório privado; na data de elaboração deste relatório, um desses pacientes tem quatro anos e desenvolvimento neurológico e psicomotor normal e os outros dois pacientes, de um e seis anos, têm atrasos leves e moderados no desenvolvimento psicomotor e neurológico, respectivamente. A tabela 1 apresenta uma comparação entre pacientes com e sem diagnóstico precoce de DXB.
Influência do diagnóstico precoce no curso da DXBa
| Diagnóstico precoce (n=12) | Diagnóstico tardio (n=71) | p | |
|---|---|---|---|
| Histórico familiar positivo (n=14/83) | 5/12 (41,6%) | 9/71 (12,6%) | 0,034 |
| Gravidade do atraso no desenvolvimento (n=58/83) | |||
| Inexistente (n=1) | 1/12 (8,3%) | 0/46 (0%) | 0,230 |
| Grave (n=15) | 2/12 (16,7%) | 13/46 (28,3%) | |
| Moderada (n=20) | 4/12 (33,3%) | 16/46 (34,8%) | |
| Leve (n=22) | 5/12 (41,7%) | 17/46 (37%) | |
| Leucina ≥ 1000μmol/L no diagnóstico (n=53/73) | 9/12 (75%) | 44/61 (72,1%) | 0,716 |
| Sintomas presentes no diagnóstico (n=80/83) | 9/12 (75%) | 71/71 (100%) | 0,02 |
| Período de diagnóstico (n=83/83) | |||
| 1992‐2001 (n=31) | 2/12 (16,7%) | 29/71 (40,8%) | 0,195 |
| 2002‐2011 (n=52) | 10/12 (83,3%) | 42/71 (59,1%) | |
| Sobrevivência em 2011 (n=68/83) | |||
| Vivos (n=48) | 9/12 (75%) | 39/56 (69,6%) | 0,866 |
| Falecidos (n=20) | 3/12 (25%) | 17/56 (30,4%) | |
As manifestações clínicas mais comuns no momento da inclusão dos pacientes eram de atraso no desenvolvimento psicomotor e neurológico (98,8%) e situação nutricional deficiente (74,7%). Dois pacientes estavam acima do peso.
A idade média no diagnóstico não foi significativamente associada à gravidade do atraso no desenvolvimento (n=58/83; p=0,31) nem a níveis elevados de leucina (n=57/73; p=0,961).
Dos pacientes, 73 (88%) tinham DXB clássica (idade média no diagnóstico: 60 dias; IQR: 27,5‐180 dias), oito (9,6%) tinham DXB moderada (idade média no diagnóstico: 257 dias; IQR: 33,7‐668 dias) e dois (2,4%) tinham DXB intermitente (idade no diagnóstico: seis e sete anos, respectivamente). Em pacientes com a forma moderada, as manifestações clínicas mais comuns no diagnóstico eram baixo apetite e anomalias respiratórias, ao passo que em pacientes com a forma intermitente essas manifestações eram convulsões e anomalias respiratórias. Todos os pacientes com as formas moderada/intermitente estavam vivos na data de elaboração deste relatório.
TratamentoDos pacientes, 58 (69,9%) estavam sendo tratados por neurologistas, 56 (67,5%) por geneticistas médicos, 49 (59,0%) por pediatras e 46 (55,4%) por nutricionistas. Outros profissionais envolvidos no tratamento e acompanhamento de pacientes incluíam neonatologistas, gastroenterologistas, nutricionistas, fonoaudiólogos e fisioterapeutas.
Os pacientes em nossa amostra receberam acompanhamento em 16 centros de tratamento, com uma média de cinco pacientes por centro (IQR: 1,75‐6,5). O uso de uma fórmula metabólica específica para a DXB foi relatado em 62 de nossos 73 pacientes (74,7%). Três pacientes haviam sido submetidos a transplante de fígado; em dois casos, o procedimento foi feito no Brasil. Dos pacientes, 37 (59,7%) receberam a fórmula metabólica regularmente (idade média: cinco anos; IQR: um‐7,5 anos); os que relataram falta de fornecimento da fórmula tinham idade média de dois anos (IQR: 0,5‐5). O tempo médio decorrido entre o diagnóstico e o recebimento da fórmula foi de 17,5 dias (IQR: 5,75‐30 dias). Não houve associação significativa entre a gravidade do atraso no desenvolvimento e a regularidade do fornecimento da fórmula (n=40/62, p=0,074).
ÓbitosDos pacientes para os quais os dados estavam disponíveis (n=68), 20 – todos com DXB clássica – vieram a óbito antes do início do estudo. A idade média no óbito era de 225 dias (IQR: 127,5‐365 dias). Não houve uma correlação estatisticamente significativa entre o desfecho fatal e os níveis de leucina no diagnóstico (p=0,568).
DiscussãoNo melhor de nosso conhecimento, este é o primeiro estudo a descrever o perfil de pacientes brasileiros com DXB. A população atual do Brasil está estimada em 190.732.694, com 2.944.928 nascidos vivos por ano.13 Portanto, considerando uma incidência média de DXB de 1:100.000 no país, são esperados aproximadamente 300 novos diagnósticos da doença em 10 anos – uma estimativa muito mais baixa do que aquela realmente incluída no estudo pela amostra. Isso sugere que a DXB seja subdiagnosticada no país.
A DXB atende à maioria dos critérios de Wilson e Jungner14 para a triagem: por exemplo, existe uma fase latente reconhecível ou sintomática precoce e um tratamento aceito para pacientes com confirmação da doença. Em países onde a DXB está incluída na triagem neonatal, os pacientes normalmente são diagnosticados antes do 10° dia de vida.3,4 Por outro lado, em países onde a DXB não está incluída nos programas públicos de triagem neonatal, como o Brasil, o diagnóstico normalmente é postergado, ocorre em idades semelhantes àquelas relatadas em nossa amostra.15,16 A predominância da DXB clássica e de pacientes sintomáticos em nossa amostra poderia também ser devida à não inclusão da DXB nos programas públicos de triagem neonatal, já que a literatura sugere que a triagem neonatal permite o diagnóstico antecipado das formas mais leves da doença.5
Como esperado, os pacientes com histórico familiar positivo foram testados antes daqueles sem histórico familiar de DXB; isso foi devido, provavelmente, ao aconselhamento genético das famílias que já tinham um filho com a doença e estavam, assim, cientes do risco de recidiva e da necessidade de investigação.
Nosso estudo constatou uma tendência crescente no número de diagnósticos de DXB na última década, o que coincidiu com o estabelecimento do Siem e a implementação de um programa nacional de triagem neonatal pelo Ministério da Saúde.17 Os motivos por trás dessa tendência são desconhecidos, porém podem refletir uma maior ciência de EIMs em geral por parte dos prestadores de serviços de saúde, bem como maior ciência de manifestações clínicas precoces dessas doenças. Contudo, o aumento não foi estatisticamente significativo e não havia diferença significativa na idade no momento do diagnóstico entre os dois períodos, o que corrobora nossa convicção de que uma parte substancial de pacientes com DXB continua a falecer sem diagnóstico ou tratamento no Brasil. Uma situação semelhante ocorre na Malásia, onde a triagem neonatal não inclui a DXB: o diagnóstico normalmente é tardio e a DXB parece menos prevalente do que o esperado.18
Atrasos psicomotor e no desenvolvimento neurológico foram detectados em praticamente todos os pacientes na amostra. Pouco mais da metade dos pacientes que receberam a fórmula metabólica para DXB relataram que foi fornecida de forma regular. Contudo, a maioria dos pacientes apresentou situação nutricional inadequada. Vale ressaltar que os pacientes com DXB sempre devem ser acompanhados por prestadores de cuidados nutricionais e apenas metade dos pacientes em nossa amostra eram acompanhados por um nutricionista.19 Contudo, a maioria dos pacientes que receberam a fórmula metabólica foi monitorada por nutricionistas (dados não apresentados). Os neurologistas eram os profissionais mais comumente responsáveis pelo acompanhamento dos pacientes, que poderá ser secundário à alta frequência de atraso no desenvolvimento nessa amostra.
No Brasil, o tempo entre o diagnóstico e o recebimento da fórmula metabólica é longo e variável. Quando os pacientes foram diagnosticados no estágio agudo da doença, durante uma internação devido a descompensação metabólica, estavam suscetíveis ao início da introdução da fórmula metabólica no diagnóstico (caso a fórmula estivesse disponível no hospital da internação, obviamente). Por outro lado, os pacientes diagnosticados em um estágio não agudo da doença e tratados de forma ambulatorial estavam propensos a receber a fórmula apenas posteriormente; na verdade, esses pacientes normalmente garantem o acesso à fórmula metabólica por meio de processos judiciais. Novamente, vale ressaltar que o uso de fórmula isenta de BCAA é essencial, de forma a garantir a quantidade de proteína necessária para o crescimento e desenvolvimento adequados.1 Recentemente, foram feitos estudos em ratos com DXB clássica e intermediária recém‐induzidas para avaliar as consequências do rápido acúmulo de BCAA e para avaliar as possíveis opções de tratamento, como norleucina.20
Os níveis de leucina no diagnóstico mostraram‐se elevados, com um valor médio de 1.693μmol/L. Os níveis de leucina superiores a 1.000μmol/L são considerados críticos, pois podem produzir dano irretratável ou até mesmo levar a óbito.3,21,22 Contudo, não houve associação significativa entre a gravidade do atraso no desenvolvimento e os níveis de leucina no diagnóstico. Isso pode ser atribuído ao fato de que o controle metabólico de longo prazo é considerado um fator determinante de desenvolvimento psicomotor e cognitivo mais decisivo do que os níveis de leucina no diagnóstico.22
Neste estudo, a vantagem do diagnóstico precoce parece não ter sido relevante devido à falta de manejo clínico de curto e longo prazos. Conforme relatado por pacientes filipinos,15 nenhum protocolo clínico para o tratamento da DXB em estágio agudo estava disponível no Brasil e os pacientes não receberam a fórmula metabólica de forma confiável. Por outro lado, em um estudo de Morton et al.4 no qual os pacientes tiveram acesso à formula e um protocolo clínico foi seguido no estágio agudo da doença, o resultado geral foi melhor e os pacientes atingiram um desenvolvimento mais adequado.
Em vista da recente adoção de uma política pública específica para o diagnóstico e tratamento de doenças raras no Brasil,23,24 sugerimos que as seguintes etapas sejam seguidas a fim de melhorar ainda mais a qualidade de vida dos pacientes com DXB no país: a) expandir o programa público de triagem neonatal para incluir a DXB entre as doenças identificadas; b) desenvolver a capacidade de fazer testes de aloisoleucina localmente; c) oferecer fórmulas metabólicas específicas a todos os pacientes, sem a necessidade de intervenção legal; d) estabelecer um centro nacional especializado em transplante de fígado para doenças metabólicas; e e) estabelecer uma rede de equipes multidisciplinares que abranja médicos, enfermeiras e nutricionistas no tratamento de erros inatos do metabolismo para o desenvolvimento de protocolos nacionais para o tratamento da DXB. A criação da Rede DXB (http://redexaropedobordo.com.br/), estabelecida em 2010 para promover a educação sobre o diagnóstico e tratamento da DXB e patrocinada por uma agência brasileira de pesquisas e pelo Ministério da Ciência e Tecnologia, é um dos primeiros passos com relação a esse objetivo.
FinanciamentoCoordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), Fundo de Incentivo à Pesquisa e Eventos (Fipe‐HCPA) e Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) – bolsa n° MCT/CNPq/CT‐Saúde 57/2010.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Aos profissionais da Rede DXB que contribuíram para este artigo, bem como à equipe do Setor de Genética Médica do Hospital de Clínicas de Porto Alegre, RS, Brasil, principalmente aos profissionais envolvidos na análise biomédica de pacientes com suspeita de doenças metabólicas.
Como citar este artigo: Herber S, Schwartz IV, Nalin T, Netto CB, Camelo Junior JS, Santos ML, et al. Maple syrup urine disease in Brazil: a panorama of the last two decades. J Pediatr (Rio J). 2015;91:292–8.
Estudo feito na Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brasil.




