



To show the general prevalence and to characterize tetrahydrobiopterin (BH4) deficiencies with hyperphenylalaninemia, identified by the Neonatal Screening Program of the State of Minas Gerais.
MethodsDescriptive study of patients with BH4 deficiency identified by the Neonatal Screening Program of the State of Minas Gerais.
ResultsThe prevalence found was 2.1 for 1,000,000 live births, with a frequency of 1.71% among hyperphenylalaninemias. There were four cases (40%) with 6-pyruvoyl-tetrahydropterin synthase deficiency, three with GTP cyclohydrolase I – autosomal recessive form deficiency, and three with dihydropteridine reductase deficiency (30% each). Six patients were diagnosed due to clinical suspicion and four cases due to systematic screening in neonatal screening. After the start of the treatment, patients identified by neonatal screening had rapid improvement and improved neuropsychomotor development compared to those diagnosed by the medical history.
ConclusionsThe prevalence of BH4 deficiencies in Minas Gerais was slightly higher than that found in the literature, but the frequency among hyperphenylalaninemias was similar. Although rare, they are severe diseases and, if left untreated, lead to developmental delays, abnormal movements, seizures, and premature death. Early treatment onset (starting before 5 months of age) showed good results in preventing intellectual disability, justifying the screening of these deficiencies in newborns with hyperphenylalaninemia identified at the neonatal screening programs for phenylketonuria.
Apresentar a prevalência geral e caracterizar as deficiências de tetrahidrobiopterina - BH4 - com hiperfenilalaninemia, identificadas pelo Programa de Triagem Neonatal do Estadode Minas Gerais.
MétodosEstudo descritivo de pacientes com deficiência de BH4 do Programa de Triagem Neonatal do Estado de Minas Gerais.
ResultadosA prevalência encontrada foi de 2,1 para 1.000.000 recém-nascidos vivos e a frequência de 1,71%, dentre as hiperfenilalaninemias. Quatro casos (40%) com deficiência de 6-piruvoil-tetrahidropterina sintase, três com deficiência de GTP ciclohidrolase I e três com deficiência de dihidropteridina redutase (30% cada um). Seis pacientes foram diagnosticadospor suspeita clínica e quatro pela pesquisa sistemática na triagem neonatal. Após o início do tratamento, os pacientes identificados pela triagem neonatal tiveram melhora rápida e melhor desenvolvimento neuropsicomotor em comparação com aqueles diagnosticados pela história clínica.
ConclusõesA prevalência das deficiências de BH4 em Minas Gerais foi um pouco maior que a encontrada na literatura, mas a frequência, entre as hiperfenilalaninemias, foi semelhante. Embora raras, são graves e, se não tratadas, levam a atraso de desenvolvimento, movimentos anormais, convulsões e morte precoce. O tratamento precoce (início antes dos 5 meses) mostrou bons resultados na prevenção de deficiência intelectual, justificando a pesquisa dessas deficiências nos recém-nascidos com hiperfenilalaninemia pelos programas de triagem neonatalpara fenilcetonúria.
As hiperfenilalaninemias são manifestações dos defeitos genéticos mais frequentemente envolvidos no metabolismo de aminoácidos.1 Até o momento são conhecidos cinco defeitos que levam a estados de hiperfenilalaninemia: na enzima fenilalanina hidroxilase, causa a fenilcetonúria, e em quatro enzimas envolvidas na síntese ou regeneração da tetrahidrobiopterina (BH4), um importante cofator de enzimas envolvidas na síntese de tirosina, dopamina, serotonina, óxido nítrico e glicerol.2
As quatro deficiências de BH4 que cursam com hiperfenilalaninemia são deficiência de GTP ciclohidrolase I – forma autossômica recessiva (GTPCH I), deficiência de 6‐piruvoil‐tetrahidropterina sintase (PTPS), deficiência de dihidropteridina redutase (DHPR) e deficiência de pterina‐4α‐carbinolamina desidratase (PCD). Representam, aproximadamente, 2% dos casos de hiperfenilalaninemia com uma prevalência mundial de 1 para 1.000.000 de nascidos vivos.3
Como a BH4 está envolvida na síntese de neurotransmissores como dopamina e serotonina, as deficiências desse cofator podem levar não só a estados de hiperfenilalaninemia, como também a sintomas e sinais neurológicos decorrentes da deficiência desses neurotransmissores.4
Uma vez que essas deficiências cursam com hiperfenilalaninemia, é possível sua identificação pelos programas de triagem neonatal para fenilcetonúria. Isso permite que o diagnóstico seja feito já nas primeiras semanas de vida5,6 e seja iniciado, prontamente, o tratamento. A rotina atual de se pesquisarem as deficiências de BH4 em todo recém‐nascido com hiperfenilalaninemia, pela análise de pterinas e atividade da enzima DHPR em amostras de sangue em papel filtro, e iniciar o tratamento precoce tem resultado em melhores níveis intelectuais em pacientes afetados do que os encontrados anteriormente a essa rotina.7
O tratamento das deficiências de BH4 deve ser individualizado em virtude da grande heterogeneidade interindividual alélica e não alélica, característica da doença.4
No Brasil, a triagem neonatal para hiperfenilalaninemias começou na década de 1980. Em 2001, foi criado o Programa Nacional de Triagem Neonatal e, desde então, a triagem neonatal para hiperfenilalaninemias foi ampliada e hoje cobre todo o país.8 No entanto, a maioria dos estados não faz a triagem sistemática para as deficiências de BH4, o que leva a atraso no diagnóstico e no início do tratamento.
Em Minas Gerais, o Programa de Triagem Neonatal (PTNMG) é coordenado pelo Núcleo de Ações e Pesquisa em Apoio Diagnóstico (Nupad) e abrange os 853 municípios. Desde a sua implantação, já triou mais de 4.300.000 recém‐nascidos. Objetiva diagnosticar e tratar precocemente anemia falciforme, fibrose cística, hipotireoidismo congênito, deficiência de biotinidase e fenilcetonúria. Desde 2006 faz, de forma sistemática, a pesquisa para as deficiências de BH4. É, até onde sabemos, o local com o maior número de pacientes em seguimento longitudinal com essas deficiências no Brasil.
Pela raridade, as deficiências de BH4 são ainda pouco conhecidas e há poucos estudos que envolvem pacientes com esse diagnóstico.7,9–11
Existem alguns sinais e sintomas descritos, mas o fenótipo, como um todo, ainda é pouco conhecido, bem como sua evolução clínica. Como em nosso país a maioria dos programas de triagem neonatal para fenilcetonúria não faz a triagem para as deficiências de BH4, conhecer a sintomatologia clínica e a evolução desses pacientes pode contribuir para uma suspeita diagnóstica mais precoce e antecipar medidas diagnósticas e terapêuticas e melhorar sua qualidade de vida. Aos parentes próximos, o aconselhamento genético reveste‐se de grande importância no planejamento familiar.
O presente estudo teve como objetivo identificar os tipos, a prevalência geral e a frequência dessas deficiências em recém‐nascidos com hiperfenilalaninemias triados pelo PTNMG, desde sua implantação, em 1993, até dezembro de 2012. Propôs descrever as diversas características clínicas da doença e permitir sua melhor compreensão.
MétodosConduziu‐se um estudo transversal e retrospectivo, aprovado pelo Comitê de Ética da Universidade Federal de Minas Gerais – UFMG (CAAE – 04096512.0.0000.5149), em que foram identificados todos os pacientes, triados pelo PETNMG, com diagnóstico de uma das formas de deficiência de BH4 que cursam com hiperfenilalaninemia. Esse diagnóstico foi feito pelos laboratórios de Bioquímica da University Children's Hospital Zürich, na Suíça, e da Faculté Libre de Medicine, na França, a partir da análise de pterinas e da atividade de DHPR em sangue e/ou urina coletados no dia da primeira consulta dos pacientes no ambulatório de fenilcetonúria do Hospital das Clínicas da UFMG e depositados em papel filtro que, após seco, foi enviado via correio.
O cálculo da prevalência geral das deficiências de BH4 foi obtido a partir da razão entre o número de casos diagnosticados, desde a implantação do Programa de Triagem até dezembro de 2012, e o número de recém‐nascidos triados no mesmo período. No cálculo da frequência das deficiências de BH4, entre as hiperfenilalaninemias, o denominador usado foi o número de casos de hiperfenilalaninemias identificados no mesmo período.
Identificados os pacientes com as deficiências, foram registradas as características clínicas e laboratoriais apresentadas por eles até dezembro de 2012, tais como atraso de desenvolvimento neuropsicomotor, hipotonia, mioclonia, espasticidade, convulsões, hiperreflexia, hipersalivação, coriza serosa, tremor fino, irritabilidade, níveis séricos de fenilalanina. O tempo decorrido entre o início do tratamento e a melhoria ou o desaparecimento dos sintomas foi fator importante na comparação entre os pacientes que tiveram diagnóstico pela suspeita clínica e os que tiveram diagnóstico pela pesquisa sistemática das deficiências de BH4.
Coleta de dadosForam consultados os bancos de dados do Núcleo de Ações e Pesquisa em Apoio Diagnóstico da Faculdade de Medicina da Universidade Federal de Minas Gerais (Nupad/FM/UFMG), serviço de referência do Programa de Triagem no Estado de Minas Gerais e do Ambulatório de Fenilcetonúria do Serviço Especial de Genética do Hospital das Clínicas da UFMG, no qual são armazenados dados de todos os pacientes triados e daqueles em acompanhamento. Também foram consultados os prontuários dos pacientes. Os pais dos pacientes com deficiência de BH4 assinaram termo de consentimento livre e esclarecido para autorizar a participação no estudo.
Critérios de inclusãoForam incluídos no estudo todos os pacientes com hiperfenilalaninemia por deficiência de BH4, confirmados por análise de pterinas e atividade de DHPR.
ResultadosEntre setembro de 1993 e dezembro de 2012, foram triados 4.684.515 recém‐nascidos em Minas Gerais, Brasil. Desses, 586 foram identificados com hiperfenilalaninemia, dos quais 10 por deficiência de BH4, oito do sexo masculino (80%).
A prevalência encontrada foi de 2,1 para cada 1.000.000 de nascidos vivos e a frequência foi de 1,71% dentre as hiperfenilalaninemias.
Todos os pacientes foram acompanhados em consultas regulares com periodicidade de dois a seis meses.
A distribuição, por tipo de deficiência, pode ser vista na tabela 1.
Três pacientes faleceram antes dos seis anos: dois com deficiência de PTPS (aos cinco e aos 10 meses) e um com deficiência de GTPCH I (aos cinco anos). Foram mortes sem causa reconhecida e ocorreram em seus domicílios.
Os seis primeiros pacientes, todos anteriores a 2006, foram, inicialmente, diagnosticados como fenilcetonúricos, mas não responderam adequadamente ao tratamento dietético, passaram a apresentar sinais clínicos sugestivos das deficiências de BH4. A partir de 2006, quando se estabeleceu a pesquisa sistemática para as deficiências de BH4, mais quatro pacientes foram identificados.
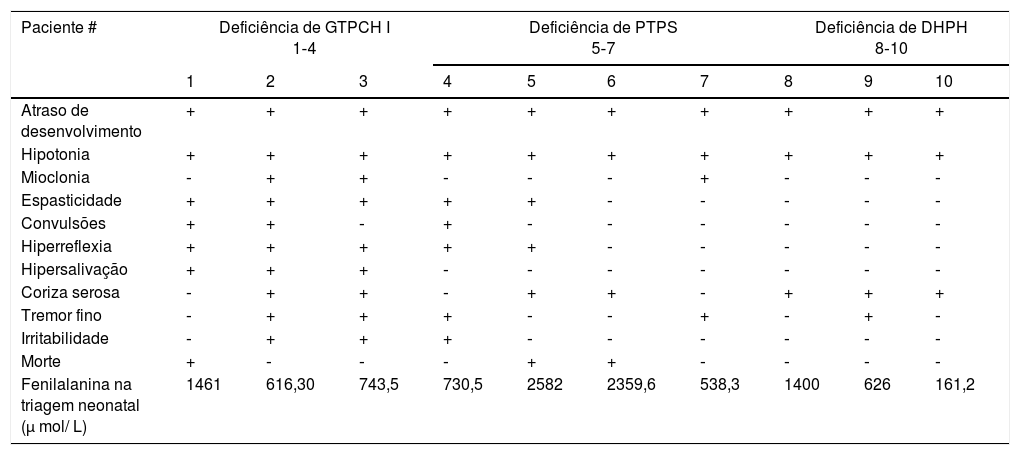
A tabela 2 resume sinais e sintomas de cada paciente, de acordo com o tipo de deficiência. Essas características clínicas surgiram antes de iniciado o tratamento e nenhuma surgiu após ele.
Sinais e sintomas encontrados nos pacientes com deficiências de BH4 em Minas Gerais: (+) sintomas presentes (‐) sintomas ausentes
| Paciente # | Deficiência de GTPCH I 1‐4 | Deficiência de PTPS 5‐7 | Deficiência de DHPH 8‐10 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Atraso de desenvolvimento | + | + | + | + | + | + | + | + | + | + |
| Hipotonia | + | + | + | + | + | + | + | + | + | + |
| Mioclonia | ‐ | + | + | ‐ | ‐ | ‐ | + | ‐ | ‐ | ‐ |
| Espasticidade | + | + | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ |
| Convulsões | + | + | ‐ | + | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hiperreflexia | + | + | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hipersalivação | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Coriza serosa | ‐ | + | + | ‐ | + | + | ‐ | + | + | + |
| Tremor fino | ‐ | + | + | + | ‐ | ‐ | + | ‐ | + | ‐ |
| Irritabilidade | ‐ | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Morte | + | ‐ | ‐ | ‐ | + | + | ‐ | ‐ | ‐ | ‐ |
| Fenilalanina na triagem neonatal (μ mol/ L) | 1461 | 616,30 | 743,5 | 730,5 | 2582 | 2359,6 | 538,3 | 1400 | 626 | 161,2 |
As tabelas 3 e 4 mostram (para pacientes com diagnóstico baseado na suspeita clínica e para pacientes com diagnóstico pela triagem neonatal respectivamente) as idades dos primeiros sintomas, do diagnóstico e do início do tratamento e do início da melhoria clinica.
Idades de início de sintomas/sinais, da confirmação diagnóstica e do início de melhoria clínica de pacientes com deficiência de BH4, identificados a partir da suspeita clínica da doença
| Deficiência de BH4 (paciente #) | Início dos sintomas | Confirmação diagnóstica e idade de início do tratamento | Início de melhoria clínica (meses após início de tratamento) |
|---|---|---|---|
| GTPCH I#1 | 2 meses | 11 meses | 4 meses (falecido aos 5 anos) |
| GTPCH I#2 | 4 meses | 9 meses | 2 a 3 meses |
| GTPCH I#3 | 2 meses (sintomas graves) | 5 meses | 2 meses |
| PTPS#4 | 3 meses | 8 anos | 5 meses |
| PTPS#5 | 4 meses | Falecido aos 5 meses | |
| PTPS#6 | 2 meses | 5 meses | 2 meses (falecido aos 10 meses) |
Idades de início de sintomas/sinais, da confirmação diagnóstica e do início de melhoria clínica de pacientes com deficiência de BH4, identificados pela triagem neonatal para a doença
| Deficiência de BH4 (paciente #) | Início dos sintomas | Confirmação diagnóstica e idade de início do tratamento | Início de melhoria clínica (meses após início do tratamento) |
|---|---|---|---|
| PTPS#7 | 3 meses | 4 meses | 1 mês |
| DHPR#8 | 4 meses | 5 meses | 1 mês |
| DHPR#9 | 2 meses | 2 meses | 1 mês |
| DHPR#10 | 2 meses | 4 meses | 1 mês |
A frequência das deficiências de BH4 apresenta grande variação regional pelo mundo. Mais da metade dos pacientes é branca (52,9%); 37,2% são turcos; 12% são árabes; 10,4% são chineses; 2,1% são indianos; 2,7% são japoneses.3 No entanto, é uma doença muito rara, corresponde, na maioria dos países, a 2% das hiperfenilalaninemias triadas.1
A descrição dos fenótipos dos diversos tipos de deficiência de BH4 é importante, pois se trata de uma doença rara, com prevalência de 1:106 e com poucas descrições de pacientes, especialmente durante longos períodos de observação.7,9–11 No Brasil, isso é mais importante, pois as análises de pterinas ainda não são feitas no país e o diagnóstico depende do envio de material biológico para o exterior. Esse fato determina que diversos estados que fazem a triagem neonatal para fenilcetonúria dependam da avaliação clínica para diagnóstico e tratamento.
Em revisão de literatura, encontramos apenas dois relatos de casos descritos no Brasil, ambos no Rio Grande do Sul, pela mesma equipe. Em 1983, Giugliani et al. descreveram dois pacientes com hiperfenilalaninemia que não evoluíam bem com o tratamento dietético para fenilcetonúria. Após análise de pterinas, o diagnóstico de deficiência de BH4 foi confirmado.12 Em 1994, Jardim et al. descreveram cinco casos de deficiências de PTPS entre pacientes com suspeita diagnóstica de erros inatos do metabolismo.13
Nosso trabalho foi feito no Ambulatório de Fenilcetonúria do Serviço Especial de Genética do Hospital das Clínicas da Universidade Federal de Minas Gerais, no qual são seguidos todos os pacientes com hiperfenilalaninemia do Estado de Minas Gerais.
Até 2006, a identificação das deficiências de BH4 era feita a partir da suspeita clínica em pacientes que não apresentavam resposta satisfatória ao tratamento dietético para fenilcetonúria, apesar da adesão. A partir daquele ano, a pesquisa para as deficiências de BH4 começou a ser feita, de forma sistemática, com dosagens de pterinas e medida da atividade da enzima DHPR de todos os pacientes triados com hiperfenilalaninemia, já na primeira consulta no Serviço. É possível que alguns tenham sido perdidos porque apresentavam, na triagem, concentrações de fenilalanina abaixo do ponto de corte usado (240μmol/L ou 4mg/dL). Entretanto, como os pacientes com hiperfenilalaninemia transitória são acompanhados por seis meses e aqueles com hiperfenilalaninemia permanente até os seis anos, as meninas retornam na puberdade14 é provável que as perdas sejam mínimas.
A partir de 2006, com a pesquisa sistemática para as deficiências de BH4 em todos os pacientes com hiperfenilalaninemia, as perdas, se ocorreram, devem ter sido ainda menores. Possíveis vieses, especialmente de investigação, podem ter decorrido devido ao pequeno número de casos encontrados. No entanto, considerando o número total de pacientes triados e sem seleção amostral, é possível que os resultados sejam representativos da população.
A prevalência encontrada foi um pouco maior do que a descrita, no entanto o percentual de pacientes, dentre as hiperfenilalaninemias, foi semelhante ao descrito na literatura.1,15,16 Em 1994, Jardim et al.,13 no Estado do Rio Grande do Sul, Brasil, estudaram pacientes com suspeita de erros inatos do metabolismo e encontraram uma prevalência muito alta de deficiência de PTPS. Isso se explica por se tratar de um estudo de pacientes selecionados com suspeita diagnóstica de doenças afins. Ainda que nossa população estudada tenha sido numerosa (4.684.515 pacientes triados) e a prevalência (2,1: 106) maior do que a descrita, o número de casos foi relativamente pequeno. Apesar disso, até onde sabemos, trata‐se do maior número de casos com seguimento longitudinal no Brasil.
De forma semelhante ao descrito na literatura,3 no presente estudo a deficiência de PTPS foi a mais comum, embora com percentual menor (40%) do que o descrito (em torno de 60%).3 Em seguida, as deficiências mais encontradas foram a deficiência de DHPR (30%) e de GTPCH I (30%). Apesar de a primeira ter apresentado frequência semelhante ao descrito,3,17 a segunda, na literatura, é apresentada com percentual bem menor (4 a 5%).3,17 Não foram identificados casos de deficiência de PCD, que a literatura relata como rara (3 a 4% dos casos).3,17
O início dos sintomas em todos os pacientes com deficiência de BH4 ocorreu entre dois e quatro meses, como descrito na literatura.1,3,17–19 Não foi observada diferença na idade de início dos sintomas nos diversos tipos.
Em oito pacientes, o nível de fenilalanina, na triagem, foi compatível com os diagnósticos de fenilcetonúria leve ou clássica. O paciente #7 teve valor de fenilalanina compatível com os diagnósticos prováveis de hiperfenilalaninemias transitória ou permanente. Já a paciente #10 teve valor de fenilalanina abaixo do valor de corte e não seria diagnosticada se a sua irmã gêmea (paciente #9) não o tivesse sido. Esses fatos alertam que as deficiências de BH4 podem ter concentrações de fenilalanina baixas à triagem neonatal e reforçam a necessidade de sua investigação em todos os pacientes, mesmo com níveis levemente aumentados de fenilalanina.20,21 Alertam, ainda, para a possibilidade de falsos negativos, mesmo quando se usa essa triagem para o diagnóstico.
Todos os sinais e sintomas surgiram antes do tratamento instituído. Todos os pacientes apresentaram atraso no desenvolvimento e hipotonia muscular, como descrito na literatura.22 O terceiro sinal mais frequente foi a coriza serosa (70% dos pacientes), seguida por tremor fino, hiperreflexia e espasticidade, presentes em 50%. Irritabilidade, mioclonias, convulsões e hipersalivação estiveram presentes em 30% dos pacientes. Esses dados, todos descritos na literatura,19,23–28 alertam que a combinação de sinais e sintomas pode ser variável e que o exame neurológico ajuda na suspeita diagnóstica.
O óbito ocorreu em 30% dos pacientes, maior do que o descrito (13%),3 evidenciou a gravidade da doença.18,29 Desses pacientes, dois tinham deficiência de PTPS e um de GTPCH I. Um dos pacientes morreu antes de ser possível iniciar a terapia. Não se sabe a causa exata do óbito de qualquer deles. Na literatura, os casos de mortes por deficiência de BH4 são atribuídos a morte súbita sem especificação exata da causa.3 Todos os pacientes que faleceram tiveram o diagnóstico feito na fase anterior ao estudo sistemático das pterinas e da atividade de DHPR.
Os pacientes com diagnóstico pós‐suspeita clínica tiveram confirmação e início do tratamento mais tardiamente, entre cinco e 11 meses. Apesar disso, o tratamento apresentou bons resultados, especialmente em relação a quadros distônicos, inclusive em um paciente com deficiência de PTPS (#4) cujo diagnóstico foi feito oito anos após o início dos sintomas. Ele apresentava grave quadro neurológico, mas hoje consegue andar, falar com dificuldade e mantém boa interação com o meio, embora ainda muito dependente para atividades diárias. Esse paciente já tinha suspeita diagnóstica desde os cinco meses, no entanto sua mãe não quis prosseguir na investigação; tinha outro filho igualmente afetado. Somente voltou ao ambulatório oito anos depois, devido à morte do filho mais velho. Esse fato alerta para importância da reação emocional da família quando não se consegue o diagnóstico precoce e o paciente não responde bem à terapêutica instituída.
A resposta ao tratamento foi um pouco mais lenta nos pacientes cujos diagnósticos foram feitos após a suspeita clínica. Esses, apesar de boa resposta clínica, permaneceram com algum grau de atraso no desenvolvimento, evidenciaram comprometimento cerebral precoce. Suspeita‐se que o desenvolvimento neuronal, a maturação funcional precoce de sinapses e a mielinização sejam alterados por deficiências de BH4 durante os períodos críticos do desenvolvimento cerebral.18
Os pacientes cujos diagnósticos foram feitos após o estudo sistemático das pterinas e da atividade de DHPR apresentaram tempo mais curto entre o início do tratamento e o início de melhoria dos sintomas (cerca de um mês). Apresentaram, também, melhor desenvolvimento neuropsicomotor, dois deles (#7 e #8) apresentaram desenvolvimento aparentemente normal até a conclusão deste estudo. Duas pacientes (#9 e #10), gêmeas, apesar de bom desenvolvimento motor, têm apresentado atraso na fala. A mãe delas teve gravidez complicada por pré‐eclâmpsia, toxoplasmose e parto pré‐termo. Devido à toxoplasmose materna, as pacientes foram tratadas para essa doença até um ano. É importante considerar a superposição da toxoplasmose e dificuldade da fala.
O tratamento precoce das deficiências de BH4 só é possível se o diagnóstico diferencial dos tipos de hiperfenilalaninemias for feito o mais brevemente possível, preferencialmente dentro do primeiro mês de vida.11 Embora não se tenha conseguido iniciar o tratamento nesse período, pois o diagnóstico ainda é feito fora do país, a evolução satisfatória dos pacientes diagnosticados justifica essa recomendação.
Os pacientes com deficiências de DHPR apresentaram a melhor evolução clínica, embora essa seja descrita como a forma mais grave de deficiência de BH4.22 O tratamento precoce provavelmente contribuiu para esse comportamento.
Nenhum paciente apresentou reações adversas aos medicamentos.
O uso da sapropterina (BH4 sintética) não foi efetuado de forma regular devido à dificuldade na obtenção da medicação em nosso meio, já que não se trata de medicação com licença de venda pela Anvisa. Somente dois pacientes (#2 e #3) com deficiência de GTPCH I fizeram o seu uso por curto tempo. O medicamento é disponível em apenas alguns países e a sua importação é muito cara. A aquisição da medicação só foi possível mediante ação judicial. Na ausência da medicação, a hiperfenilalaninemia foi controlada com dieta específica, com bons resultados.
A todos os pais foi oferecido aconselhamento genético.
O número de casos encontrados foi maior do que o esperado, mas ainda pequeno para conclusões mais definitivas. O tamanho da amostra pode explicar as dissonâncias com os percentuais descritos na literatura. Este trabalho reforça a opinião do Grupo Europeu de Fenilcetonúria, de março de 2011, de que, após a triagem neonatal para fenilcetonúria, todo recém‐nascido, mesmo com níveis séricos levemente elevados de fenilalanina, deve ser triado para deficiência de BH4.20 Essa é a recomendação atual daquele grupo aos serviços de triagem neonatal, corroborada por Han et al., que enfatizam que o diagnóstico e o tratamento precoces influenciam no prognóstico da doença e ajudam a maximizar o potencial de desenvolvimento.30 Realça, ainda, a necessidade de esses exames serem feitos em nosso país, o que permitirá diagnóstico e aconselhamento genéticos ainda mais precoces.
Como são doenças muito raras, muitos profissionais de saúde desconhecem suas características e suas formas de tratamento. Esperamos que este trabalho contribua para uma melhor compreensão dos aspectos clínicos e epidemiológicos das deficiências de BH4 no Estado de Minas Gerais.
Os resultados encontrados foram consistentes com a literatura científica. Mutações específicas já foram descritas em pequeno número de casos, mas as relações genótipo‐fenótipo ainda não são consistentes. Essa pode ser um a linha interessante de futuras pesquisas também em nosso meio.
Esperamos, no futuro próximo, poder fazer no Brasil os exames que identificam as deficiências de BH4, o que permitirá diagnósticos e tratamentos ainda mais precoces com melhoria importante na evolução clínica dos pacientes afetados.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Souza CA, Alves MR, Soares RD, Kanufre V, Rodrigues VM, Norton RC, et al. BH4 deficiency identified in a neonatal screening program for hyperphenylalaninemia. J Pediatr (Rio J). 2018;94:170–176.
Trabalho vinculado à Universidade Federal de Minas Gerais (UFMG), Hospital das Clínicas, Núcleo de Ações e Pesquisa em Apoio Diagnóstico (Nupad), Belo Horizonte, MG, Brasil.




